Scalable Synthesis of Quinazoline Derivatives for Next-Generation PAK4 Targeted Therapy
Scalable Synthesis of Quinazoline Derivatives for Next-Generation PAK4 Targeted Therapy
The global pharmaceutical landscape is increasingly focused on the development of highly selective protein kinase inhibitors for oncology applications, with Patent CN111072640A representing a significant advancement in this domain. This patent discloses a novel class of quinazoline derivatives, specifically (R)-(4-((1H-pyrazol-3-yl)amino)quinazoline-2-yl)-(N-carbamate piperazine-1-yl)-ketone derivatives, which function as potent prodrugs for PAK4 inhibition. Unlike traditional small molecule inhibitors that often suffer from poor oral bioavailability or rapid metabolic clearance, these compounds are engineered to release the active prototype drug rapidly under the action of hydrolases in vivo. This strategic design not only enhances therapeutic efficacy against tumors driven by PAK4 overexpression but also mitigates potential systemic toxicity. As a leading reliable pharmaceutical intermediate supplier, we recognize the immense potential of this chemical scaffold in addressing the unmet medical needs of patients with solid tumors such as breast, pancreatic, and colon cancers.
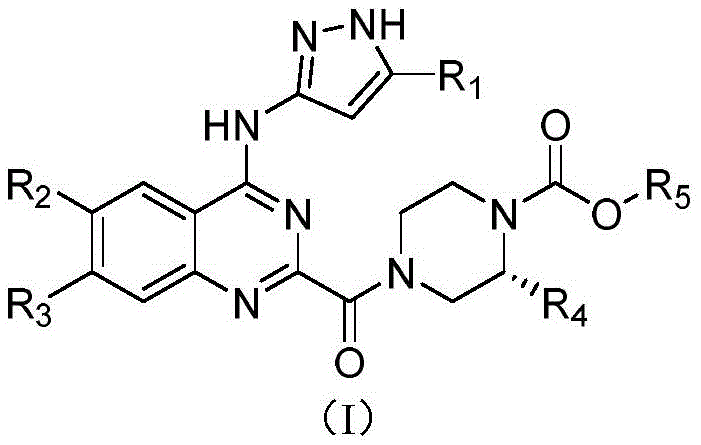
The structural versatility of Formula I allows for extensive optimization of physicochemical properties through the modulation of R groups, particularly the carbamate moiety at the piperazine nitrogen. This modification is critical for tuning the hydrolysis rate and ensuring that the active drug is released at the target site rather than in the gastrointestinal tract. The patent highlights that these derivatives exhibit superior PAK4/PAK1 selectivity, a common challenge in kinase inhibitor development due to the high homology of ATP-binding pockets across the kinase family. By leveraging this intellectual property, research and development teams can accelerate the discovery of next-generation antineoplastic agents that overcome the resistance mechanisms often observed with first-generation therapies. Our expertise in complex heterocyclic synthesis positions us to support the commercial translation of these promising candidates from benchtop discovery to industrial production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to synthesizing quinazoline-based kinase inhibitors often rely on harsh reaction conditions that can compromise the integrity of sensitive functional groups, leading to complex impurity profiles and difficult purification processes. Conventional methods frequently utilize direct chlorination followed by nucleophilic substitution, which can result in regioisomeric mixtures that are challenging to separate on a large scale. Furthermore, many existing PAK inhibitors lack a prodrug strategy, resulting in suboptimal pharmacokinetic profiles where the active compound is metabolized too quickly or fails to reach therapeutic concentrations in tumor tissue. The reliance on expensive transition metal catalysts in some cross-coupling routes also introduces the risk of heavy metal contamination, necessitating costly removal steps that impact the overall cost reduction in API manufacturing. These limitations underscore the need for a more robust, scalable, and chemically elegant synthetic pathway.
The Novel Approach
The methodology outlined in Patent CN111072640A offers a transformative solution by employing a stepwise construction of the quinazoline core that ensures high regioselectivity and purity. The novel approach utilizes a triphosgene-mediated acylation followed by a controlled cyclization with sodium ethoxide, which effectively builds the heterocyclic system under relatively mild conditions. A key innovation is the late-stage introduction of the piperazine and aminopyrazole fragments, allowing for modular diversification of the R groups without affecting the core stability. This convergent synthesis strategy significantly simplifies the process flow, reducing the number of isolation steps and minimizing solvent consumption. By incorporating a cleavable carbamate linker, the final compounds achieve a balance between stability for formulation and lability for in vivo activation, addressing the bioavailability issues that plagued earlier generations of kinase inhibitors like PF-3758309.
Mechanistic Insights into Triphosgene-Mediated Cyclization and Substitution
The core of this synthetic technology lies in the precise control of reactivity during the formation of the quinazoline ring system. The process initiates with the activation of 2-amino-5-chlorobenzoic acid using triphosgene in anhydrous tetrahydrofuran, generating a reactive acyl chloride species in situ that immediately reacts with ammonia to form the benzamide intermediate. This avoids the handling of hazardous gaseous phosgene while maintaining high atom economy. Subsequent reaction with ethyl oxalyl chloride introduces the two-carbon unit required for ring closure. The cyclization step, mediated by sodium ethoxide in ethanol, proceeds through an intramolecular nucleophilic attack of the amide nitrogen onto the ester carbonyl, followed by dehydration to aromatize the quinazoline ring. This sequence is highly efficient, typically yielding the hydroxy-quinazoline intermediate in yields exceeding 85%, demonstrating the robustness of the chemistry for scale-up.
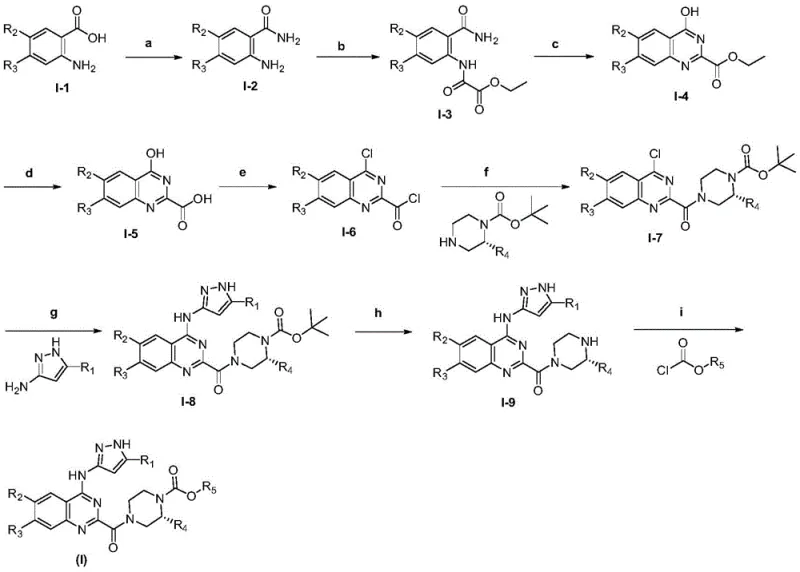
Following the construction of the core, the installation of the pharmacophore involves a nucleophilic aromatic substitution (SnAr) where the chloro group at the 4-position is displaced by a substituted 3-aminopyrazole. This step is facilitated by the electron-withdrawing nature of the quinazoline ring, which activates the 4-position towards nucleophilic attack. The use of potassium iodide as a catalyst in DMF enhances the reaction rate and ensures complete conversion, minimizing the formation of des-chloro impurities. Finally, the piperazine moiety is attached via an amide bond formation at the 2-position, followed by carbamate protection. This final capping step is crucial for the prodrug functionality, as the steric and electronic properties of the carbamate group dictate the hydrolysis kinetics. The entire pathway is designed to minimize side reactions, ensuring a clean impurity profile that meets stringent regulatory standards for high-purity pharmaceutical intermediates.
How to Synthesize Quinazoline Derivatives Efficiently
The synthesis of these complex quinazoline derivatives requires careful attention to reaction parameters such as temperature, stoichiometry, and solvent quality to ensure reproducible results. The patented procedure provides a detailed roadmap for constructing the molecule from readily available starting materials, emphasizing the importance of anhydrous conditions during the acylation and chlorination steps to prevent hydrolysis of reactive intermediates. Purification is primarily achieved through silica gel column chromatography in the laboratory examples, though for commercial scale-up, crystallization techniques would be optimized to reduce costs and environmental impact. The following guide summarizes the critical operational stages defined in the patent, serving as a foundation for process development teams aiming to replicate or adapt this chemistry for larger batches.
- Acylation of 2-amino-5-chlorobenzoic acid using triphosgene to form the benzamide intermediate.
- Cyclization with ethyl oxalyl chloride and sodium ethoxide to construct the quinazoline core.
- Final coupling with substituted aminopyrazoles and carbamates to install the pharmacophore and prodrug moiety.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the synthetic route described in this patent offers substantial advantages in terms of raw material accessibility and process safety. The starting materials, including 2-amino-5-chlorobenzoic acid and various aminopyrazoles, are commodity chemicals available from multiple global suppliers, which mitigates the risk of supply chain disruptions associated with bespoke or proprietary reagents. The avoidance of cryogenic conditions (most reactions proceed at room temperature or mild reflux) reduces energy consumption and simplifies the engineering requirements for reactor infrastructure. Furthermore, the use of common organic solvents such as THF, ethanol, and dichloromethane facilitates solvent recovery and recycling, aligning with green chemistry principles and reducing waste disposal costs. These factors collectively contribute to a more resilient and cost-effective manufacturing process.
- Cost Reduction in Manufacturing: The streamlined synthetic sequence minimizes the number of unit operations, directly translating to lower labor and equipment utilization costs. By eliminating the need for expensive transition metal catalysts often used in C-N bond formation, the process avoids the costly and time-consuming steps required for heavy metal scavenging and validation. The high yields reported in the patent examples suggest that material throughput will be efficient, reducing the cost of goods sold (COGS) per kilogram of active pharmaceutical ingredient. Additionally, the modular nature of the synthesis allows for the parallel production of different analogues using the same core intermediate, maximizing asset utilization and flexibility in response to market demand.
- Enhanced Supply Chain Reliability: The reliance on stable, shelf-stable reagents like triphosgene (as a solid alternative to phosgene) and Boc-protected piperazines ensures that inventory management is straightforward and less prone to degradation issues. The robustness of the reaction conditions means that the process is less sensitive to minor fluctuations in utility supplies, such as cooling water temperature, which enhances batch-to-batch consistency. This reliability is critical for maintaining continuous supply to downstream formulation partners, especially in the fast-paced oncology sector where clinical timelines are aggressive. The ability to source key building blocks from established chemical vendors further secures the supply chain against geopolitical or logistical shocks.
- Scalability and Environmental Compliance: The chemistry is inherently scalable, as demonstrated by the successful execution of multi-gram preparations in the patent examples without loss of efficiency. The absence of exotic or highly hazardous reagents simplifies the permitting process for new manufacturing facilities and reduces the regulatory burden associated with environmental health and safety (EHS) compliance. Waste streams are primarily composed of standard organic solvents and inorganic salts, which can be treated using conventional wastewater treatment protocols. This environmental compatibility not only lowers compliance costs but also enhances the sustainability profile of the final drug product, a growing priority for pharmaceutical buyers and investors alike.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these quinazoline derivatives. The answers are derived directly from the experimental data and technical specifications provided in Patent CN111072640A, ensuring accuracy and relevance for stakeholders evaluating this technology. Understanding these details is essential for making informed decisions about licensing, partnership, or procurement strategies.
Q: What is the primary advantage of the N-carbamate piperazine design in these quinazoline derivatives?
A: The N-carbamate piperazine design functions as a prodrug strategy, allowing the compound to remain stable during storage and administration while rapidly releasing the active prototype drug upon exposure to hydrolases in vivo, thereby improving bioavailability and reducing gastrointestinal toxicity.
Q: Are the starting materials for this synthesis commercially available?
A: Yes, the key starting materials such as 2-amino-5-chlorobenzoic acid and various substituted aminopyrazoles are readily available from major reagent suppliers, ensuring a robust and uninterrupted supply chain for large-scale manufacturing.
Q: How does this synthetic route address the selectivity challenges of PAK4 inhibitors?
A: The specific substitution pattern at the 4-position (aminopyrazole) and the 2-position (piperazine carbonyl) of the quinazoline core is engineered to maximize binding affinity for the PAK4 kinase domain while minimizing off-target effects on other kinases like PAK1.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Quinazoline Derivatives Supplier
At NINGBO INNO PHARMCHEM, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, making us the ideal partner for bringing these PAK4 inhibitors to market. Our state-of-the-art facilities are equipped to handle the specific solvent systems and reaction conditions required for quinazoline synthesis, ensuring that stringent purity specifications are met for every batch. With rigorous QC labs and a dedicated team of process chemists, we can rapidly optimize the crystallization and purification steps to deliver high-purity quinazoline derivatives that exceed industry standards. Our commitment to quality and consistency ensures that your clinical and commercial supply needs are met without interruption.
We invite you to engage with our technical procurement team to discuss how we can support your drug development pipeline with a Customized Cost-Saving Analysis tailored to your specific volume requirements. By partnering with us, you gain access to specific COA data and route feasibility assessments that validate the industrial viability of this synthetic pathway. Whether you require preclinical quantities for toxicology studies or metric tons for commercial launch, our flexible manufacturing capabilities and deep technical expertise ensure a seamless transition from development to production. Contact us today to secure a reliable supply of these critical oncology intermediates.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →