Scalable Production of High-Purity Aryl Acridines via Novel Palladium Catalysis
Scalable Production of High-Purity Aryl Acridines via Novel Palladium Catalysis
The pharmaceutical and fine chemical industries are constantly seeking robust, scalable, and cost-effective pathways for synthesizing complex nitrogen-containing heterocycles, particularly aryl acridine derivatives which serve as critical scaffolds in anticancer and antimicrobial drug development. A recent technological breakthrough documented in patent CN116283768A introduces a highly efficient four-step preparation method that addresses longstanding challenges in yield, safety, and raw material costs. This novel approach leverages a strategic combination of nucleophilic aromatic substitution and palladium-catalyzed cross-coupling reactions to construct the acridine core with exceptional precision. By utilizing accessible starting materials like o-fluorobenzonitrile and aniline, the process circumvents the need for hazardous oxygen atmospheres and expensive noble metal catalysts often required in conventional oxidative cyclization methods. For R&D directors and procurement managers alike, this represents a significant opportunity to optimize the supply chain for high-purity pharmaceutical intermediates while drastically reducing the environmental footprint of production.
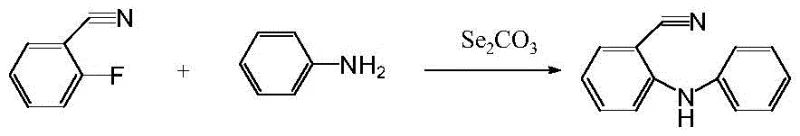
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of aryl acridine compounds has been plagued by significant operational inefficiencies and safety concerns that hinder large-scale commercialization. Traditional methodologies often rely on the condensation of o-aminoacetophenone and cyclohexanone using noble metal catalysts in the presence of trifluoroacetic acid and TBHP additives under an oxygen atmosphere. This requirement for an oxygen-rich environment introduces substantial safety risks, particularly when scaling up to multi-kilogram batches where exothermic runaway reactions become a genuine threat. Furthermore, these oxidative processes frequently suffer from low yields and incomplete conversion, leading to complex reaction mixtures that are notoriously difficult to separate and purify. Another common legacy route involves the reaction of diphenylamine with aryl formic acid under harsh acidic conditions with zinc chloride, which similarly results in poor product isolation and significant waste generation. These factors collectively drive up the cost of goods sold (COGS) and create bottlenecks for reliable aryl acridine supplier networks trying to meet the rigorous quality standards of the global pharmaceutical market.
The Novel Approach
In stark contrast to these cumbersome legacy techniques, the methodology outlined in CN116283768A offers a streamlined, modular synthesis that prioritizes both safety and economic efficiency. The process initiates with a mild nucleophilic substitution between o-fluorobenzonitrile and aniline, catalyzed by cesium carbonate in toluene at a manageable temperature of 115°C. This first step generates the key intermediate, 2-anilinobenzonitrile, with a remarkable conversion rate of 95% and LC purity exceeding 98%, effectively eliminating the purification headaches associated with older routes. The subsequent steps involve the activation of an aryl phenol, such as p-cresol, into a triflate ester, followed by a Miyaura borylation to install the necessary boron functionality. This modular design allows for the independent optimization of each fragment before the final assembly, ensuring that impurities do not carry through to the final product. By avoiding hazardous oxidants and utilizing stable, commercially available reagents, this novel approach provides a secure foundation for cost reduction in pharmaceutical intermediates manufacturing.
Mechanistic Insights into Palladium-Catalyzed Cyclization
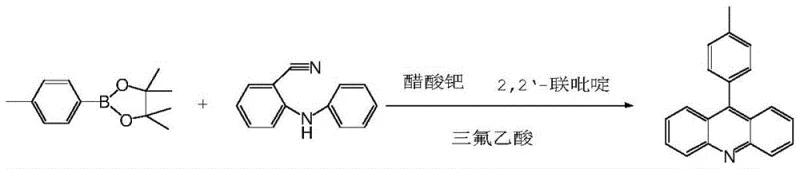
The cornerstone of this synthetic strategy is the final intramolecular cyclization step, which elegantly constructs the rigid acridine framework through a palladium-catalyzed C-N and C-C bond formation sequence. In this critical transformation, the 2-anilinobenzonitrile intermediate reacts with the arylboronic acid pinacol ester in the presence of palladium acetate, 2,2'-bipyridine, and trifluoroacetic acid (TFA). The mechanism likely proceeds through an oxidative addition of the palladium catalyst into the nitrile-activated system or via a directed C-H activation pathway facilitated by the aniline nitrogen, followed by transmetallation with the boron species. The presence of TFA plays a dual role: it acts as a proton source to facilitate the reductive elimination step and helps solubilize the polar intermediates, ensuring a homogeneous reaction environment. This precise control over the catalytic cycle minimizes side reactions such as homocoupling of the boronic ester or hydrolysis of the nitrile group, which are common pitfalls in similar cross-coupling reactions. The result is a highly selective formation of the aryl acridine core with minimal byproduct formation, demonstrating the sophistication of modern transition metal catalysis in complex molecule synthesis.
Beyond the primary catalytic cycle, the process incorporates robust impurity control mechanisms inherent to the choice of reagents and workup procedures. The use of pinacol boronic esters rather than free boronic acids enhances stability and reduces the formation of boroxine byproducts, which can complicate downstream processing. Furthermore, the reaction conditions are tuned to ensure that the palladium catalyst remains active throughout the 12-hour reflux period without decomposing into inactive black palladium precipitates, a common issue that leads to batch failures. Post-reaction workup involves a simple aqueous wash to remove inorganic salts and residual acids, followed by concentration to dryness, yielding the final product with an LC purity of greater than 99%. This high level of purity is achieved without the need for column chromatography, relying instead on the intrinsic selectivity of the catalytic system and the crystallization properties of the product. For quality assurance teams, this translates to a consistent impurity profile that is easier to characterize and control, meeting the stringent specifications required for API intermediates.
How to Synthesize Aryl Acridine Efficiently
The synthesis protocol described in the patent provides a clear, reproducible roadmap for producing aryl acridine derivatives with high fidelity. The procedure is divided into four distinct stages, each optimized for maximum yield and minimal waste. The initial coupling reaction sets the stage by establishing the aniline linkage, followed by the activation of the phenolic component into a reactive triflate. The third stage introduces the boron handle necessary for the final coupling, utilizing a standard Miyaura borylation condition with Pd2dba3 and X-PHOS ligands to ensure high turnover. Finally, the cyclization step brings the fragments together under acidic conditions to close the ring. Detailed standardized synthesis steps are provided below to guide process chemists in replicating this high-efficiency route.
- Perform coupling reaction of o-fluorobenzonitrile and aniline under alkali catalysis (Cs2CO3) at 115°C to generate 2-anilino-benzonitrile.
- React aryl phenol (e.g., p-cresol) with trifluoromethanesulfonic anhydride and triethylamine to generate aryl trifluoromethanesulfonate.
- Convert the aryl triflate to arylboronic acid pinacol ester using bis(pinacolato)diboron and Pd catalyst.
- Cyclize 2-anilino-benzonitrile and the arylboronic ester using Pd(OAc)2, 2,2'-bipyridine, and TFA to form the final aryl acridine.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented methodology offers compelling advantages that directly address the pain points of procurement managers and supply chain directors. The shift away from hazardous oxygen atmospheres and expensive noble metal catalysts in the early stages of synthesis significantly de-risks the manufacturing process. By utilizing commodity chemicals like aniline, o-fluorobenzonitrile, and p-cresol, the reliance on specialized, high-cost starting materials is minimized, creating a more resilient supply chain that is less susceptible to market volatility. The simplicity of the workup procedures, which primarily involve aqueous washes and filtration rather than complex distillations or chromatographic separations, further contributes to operational efficiency. This streamlined workflow reduces the demand on plant equipment and labor hours, allowing for faster batch turnover and improved capacity utilization. Consequently, this technology enables substantial cost savings in the production of high-value heterocyclic intermediates.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven by the replacement of expensive oxidants and noble metal catalysts with more affordable alternatives like cesium carbonate and catalytic palladium systems. The high conversion rates observed in each step, particularly the 95% yield in the first step and 90% in the final cyclization, mean that less raw material is wasted, directly lowering the material cost per kilogram of finished product. Additionally, the elimination of complex purification steps reduces solvent consumption and waste disposal costs, contributing to a leaner manufacturing budget. These factors combine to offer a significantly reduced cost structure compared to traditional oxidative cyclization methods.
- Enhanced Supply Chain Reliability: The reliance on widely available bulk chemicals ensures that production schedules are not disrupted by shortages of exotic reagents. Starting materials such as toluene, triethylamine, and potassium acetate are standard inventory items for most chemical manufacturers, simplifying procurement logistics. The robustness of the reaction conditions, which tolerate standard reflux temperatures and atmospheric pressure, means that the process can be executed in general-purpose reactors without requiring specialized high-pressure or cryogenic equipment. This flexibility enhances the overall reliability of the supply chain, ensuring consistent delivery of high-purity aryl acridines to downstream customers.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, with examples demonstrating successful translation from gram to multi-kilogram scales without loss of efficiency. The absence of hazardous oxygen atmospheres removes a major safety barrier to scaling, while the aqueous workup minimizes the generation of organic waste streams. The high atom economy of the coupling reactions and the ability to recover and recycle solvents like toluene further align the process with green chemistry principles. This makes the technology not only commercially viable but also environmentally sustainable, meeting the increasingly strict regulatory requirements for chemical manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this aryl acridine synthesis technology. These insights are derived directly from the experimental data and process descriptions found in the patent literature, providing a factual basis for decision-making. Understanding these details is crucial for evaluating the feasibility of adopting this route for commercial production.
Q: What are the primary cost advantages of this aryl acridine synthesis method?
A: The method utilizes inexpensive and readily available raw materials such as o-fluorobenzonitrile, aniline, and cresol, avoiding the need for expensive noble metal catalysts in the initial steps and eliminating the requirement for hazardous oxygen atmospheres found in conventional methods.
Q: How does this process improve product purity compared to traditional routes?
A: The novel route achieves a main product conversion rate of up to 95% with LC purity exceeding 99% in the final step, significantly reducing the burden of downstream purification and avoiding the difficult separation issues associated with incomplete reactions in older methodologies.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the process features simple post-treatment operations such as aqueous washing and filtration without complex chromatography, and operates under standard reflux conditions, making it highly scalable and safe for commercial manufacturing environments.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Aryl Acridine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this patented synthesis route for the production of high-performance pharmaceutical intermediates. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive a consistent and reliable supply of critical materials. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, including the >99% LC purity achievable through this novel method. We are committed to leveraging advanced catalytic technologies to deliver superior value to our global partners, bridging the gap between innovative academic research and industrial reality.
We invite pharmaceutical companies and research institutions to collaborate with us to explore the full capabilities of this aryl acridine synthesis platform. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to obtain specific COA data and route feasibility assessments that will demonstrate how we can support your drug development pipeline with cost-effective, high-quality intermediates.