Optimizing PARP Inhibitor Production: A Novel Salt Formation Strategy for Industrial Scale
Introduction to Advanced PARP Inhibitor Intermediate Manufacturing
The landscape of oncology drug development has been significantly transformed by the advent of Poly(ADP-ribose) polymerase (PARP) inhibitors, which exploit synthetic lethality in tumor cells with defective DNA repair mechanisms. As demand for these potent therapeutics grows, the chemical industry faces the critical challenge of supplying high-quality intermediates at a commercial scale. Patent CN111278830B discloses a groundbreaking preparation method for a key PARP inhibitor intermediate, addressing long-standing bottlenecks in purity and yield that have plagued traditional synthesis routes. This technical insight report analyzes the novel salt formation strategy detailed in the patent, offering a robust solution for reliable PARP inhibitor intermediate suppliers aiming to optimize their production pipelines. By shifting from complex chromatographic separations to efficient crystallization techniques, this methodology ensures the delivery of high-purity pharmaceutical intermediates essential for downstream API manufacturing.
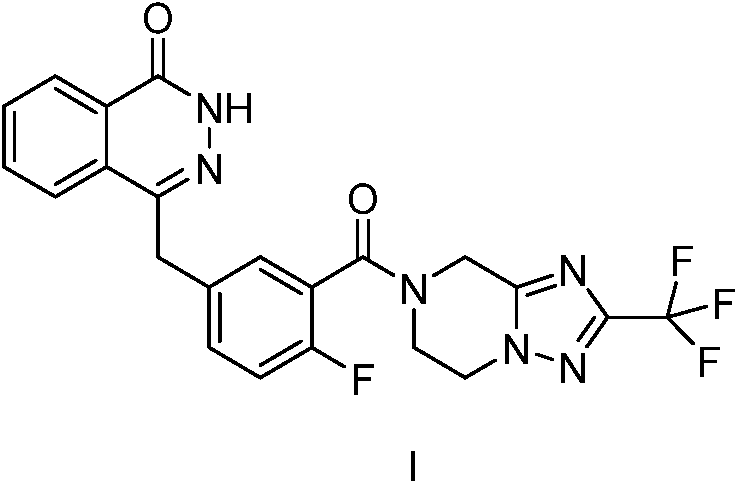
The core innovation lies in the manipulation of the triazole-piperazine scaffold, a structural motif common in many kinase and PARP inhibitors. The patent specifically targets the purification of the intermediate represented by Formula II, which serves as the nucleophilic partner in the final amide coupling reaction to form the active drug substance (Formula I). Historically, the synthesis of such nitrogen-rich heterocycles has been fraught with difficulties regarding the removal of polar impurities and residual catalysts. The disclosed method overcomes these hurdles by introducing a strategic acid-addition salt formation step, converting the free base into a stable, crystalline solid (Formula II'). This transformation not only simplifies isolation but also dramatically enhances the chemical purity of the intermediate, thereby safeguarding the quality of the final PARP inhibitor product against carryover impurities that could compromise patient safety or regulatory approval.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies, such as those described in WO2004032836, have traditionally relied on labor-intensive and inefficient purification techniques for triazole-piperazine intermediates. A common approach involves the use of preparative column chromatography, which, while effective on a laboratory scale, is economically and environmentally unsustainable for industrial production. The excessive consumption of organic solvents required for elution generates substantial hazardous waste, driving up disposal costs and complicating environmental compliance. Furthermore, chromatographic separation often fails to completely remove structurally similar by-products, leading to intermediates that require further processing. Another prevalent strategy involves the use of protecting groups, such as BOC (tert-butyloxycarbonyl), to mask the reactive imino functionality during synthesis. While this protects the amine, it introduces additional synthetic steps for protection and deprotection, inevitably lowering the overall process yield and increasing the manufacturing lead time. Additionally, the hydrogenation steps typically employed to generate the piperazine ring often utilize palladium catalysts, which are notoriously difficult to remove completely from the final product without specialized scavenging resins, posing a risk of heavy metal contamination in the final API.
The Novel Approach
In stark contrast to these cumbersome legacy processes, the method disclosed in CN111278830B introduces a streamlined purification protocol centered on salt formation. Instead of struggling to purify the free base oil or solid via chromatography, the process converts the crude intermediate directly into an acid addition salt. This simple chemical modification fundamentally alters the physical properties of the molecule, transforming it into a crystalline solid that can be easily isolated via filtration. The patent demonstrates that treating the crude reaction mixture with acids such as hydrochloric acid, phosphoric acid, or maleic acid results in the precipitation of the salt form (Formula II'), leaving soluble impurities in the mother liquor. This approach effectively bypasses the need for column chromatography and eliminates the extra steps associated with protecting group chemistry. The result is a significantly shorter synthetic route with fewer unit operations, which translates directly to reduced operational expenditure and a smaller environmental footprint. Moreover, the crystallization process inherently excludes many non-isomorphous impurities, including residual palladium species, ensuring a higher quality input for the subsequent coupling reaction.
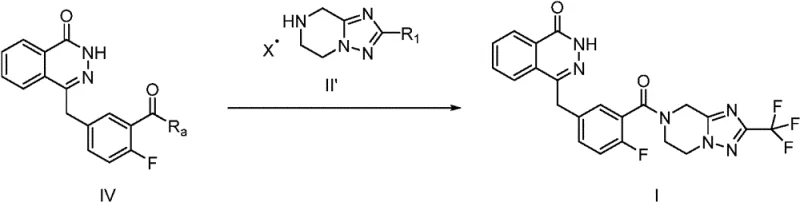
Mechanistic Insights into Salt-Mediated Purification and Hydrogenation
The chemical elegance of this process lies in the interplay between catalytic hydrogenation and acid-base chemistry. The synthesis begins with the reduction of an unsaturated precursor (Formula III) to generate the saturated piperazine-triazole system. This hydrogenation is typically conducted using a heterogeneous catalyst like palladium on carbon (Pd/C) in a protic solvent such as methanol. The mechanism involves the adsorption of hydrogen gas and the substrate onto the metal surface, facilitating the addition of hydrogen across the double bond. Once the reduction is complete, the catalyst is removed by filtration. However, the resulting free base (Formula II) is often prone to oxidation or may exist as an oil that traps solvent and impurities. The critical innovation occurs in the subsequent step where the free base is treated with a stoichiometric amount of acid. The basic nitrogen atoms in the piperazine or triazole rings accept protons to form cationic species, which then pair with the acid anions to create an ionic lattice. This lattice structure promotes orderly crystal growth, which is the thermodynamic driver for high purity. Impurities that do not fit into this crystal lattice are excluded and remain in the solution phase.
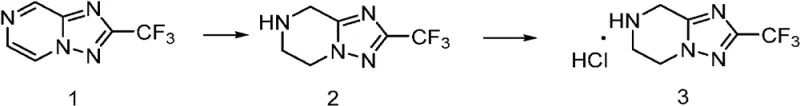
Furthermore, the choice of acid plays a pivotal role in the efficiency of impurity control. The patent data indicates that hydrochloric acid is particularly effective, yielding solids with HPLC purity exceeding 99.6%. The formation of the hydrochloride salt likely creates a highly stable crystalline form that is less hygroscopic and more chemically robust than the free base. This stability is crucial for storage and transportation, reducing the risk of degradation before the intermediate is used in the next manufacturing step. From a mechanistic standpoint, the protonation also deactivates the nucleophilicity of the amine temporarily, preventing unwanted side reactions during workup. The ability to recrystallize or slurry the salt form provides an additional layer of purification, allowing manufacturers to tune the particle size distribution and polymorphic form if necessary. This level of control over the solid-state properties is unattainable with amorphous oils produced by conventional chromatographic methods, making the salt formation strategy superior for ensuring batch-to-batch consistency in a GMP environment.
How to Synthesize High-Purity PARP Inhibitor Intermediate Efficiently
Implementing this novel synthesis route requires careful attention to reaction parameters to maximize the benefits of the salt formation technique. The process is designed to be scalable, moving seamlessly from kilogram to multi-ton production without the linear increase in complexity seen with chromatographic methods. Operators must ensure complete hydrogenation before initiating the salt formation step to avoid carrying over unsaturated impurities that could co-crystallize. The selection of solvent for the salt formation is also critical; the patent suggests using ethyl acetate or isopropanol, which offer a good balance of solubility for the free base and insolubility for the resulting salt. Below is a summary of the standardized operational logic derived from the patent examples, detailing the critical control points for successful execution.
- Perform catalytic hydrogenation of the unsaturated precursor (Formula III) using Pd/C in methanol to generate the free base intermediate.
- Concentrate the reaction mixture and dissolve the residue in a suitable solvent like ethyl acetate.
- Add an acid solution (e.g., HCl in ethyl acetate) to adjust pH to 2-3, inducing precipitation of the pure salt form (Formula II').
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this salt-based purification technology offers compelling economic and logistical advantages over traditional methods. The primary value driver is the drastic simplification of the downstream processing workflow. By eliminating column chromatography, manufacturers remove a major bottleneck that typically limits throughput and consumes vast quantities of expensive organic solvents. This reduction in solvent usage not only lowers raw material costs but also significantly decreases the volume of hazardous waste requiring treatment, leading to substantial cost savings in environmental compliance and waste disposal. Furthermore, the removal of protecting group strategies reduces the number of reaction steps, which directly correlates to lower labor costs, reduced equipment occupancy time, and higher overall plant capacity. These efficiencies allow suppliers to offer more competitive pricing for high-purity pharmaceutical intermediates while maintaining healthy margins.
- Cost Reduction in Manufacturing: The elimination of chromatographic purification and protecting group chemistry fundamentally alters the cost structure of the intermediate. Traditional methods incur high costs due to the purchase of silica gel, large volumes of HPLC-grade solvents, and the energy required for solvent recovery. In contrast, the salt formation method relies on inexpensive inorganic acids and standard crystallization equipment. The high yields reported in the patent examples, often exceeding 90%, mean that less starting material is wasted, further driving down the cost of goods sold (COGS). Additionally, the simplified process reduces the risk of batch failures associated with complex separations, ensuring a more predictable and stable cost base for long-term supply agreements.
- Enhanced Supply Chain Reliability: Supply chain resilience is heavily dependent on the robustness of the manufacturing process. The novel method's reliance on standard unit operations like filtration and drying makes it less susceptible to the variability that often plagues chromatographic separations. The ability to produce a stable solid intermediate improves inventory management, as crystalline salts generally have better shelf-life and storage characteristics than oils or unstable free bases. This stability reduces the risk of supply disruptions caused by material degradation during transit or storage. Moreover, the shorter cycle time per batch allows manufacturers to respond more agilely to fluctuations in market demand, ensuring that reliable PARP inhibitor intermediate suppliers can meet tight delivery schedules without compromising on quality.
- Scalability and Environmental Compliance: Scaling up chromatographic processes is notoriously difficult and often requires disproportionate increases in facility footprint and solvent handling infrastructure. The salt formation approach scales linearly and efficiently, fitting comfortably within existing reactor trains without the need for specialized chromatography columns. This ease of scale-up facilitates the commercial scale-up of complex pharmaceutical intermediates from pilot plant to full commercial production. From an environmental perspective, the reduction in solvent intensity aligns with green chemistry principles, helping pharmaceutical companies meet their sustainability goals. The process minimizes the generation of mixed solvent waste streams, simplifying waste treatment and reducing the overall carbon footprint of the manufacturing operation.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis route. These insights are derived directly from the experimental data and claims within CN111278830B, providing clarity on the practical aspects of adopting this technology. Understanding these details is crucial for technical teams evaluating the feasibility of integrating this intermediate into their existing API manufacturing workflows.
Q: Why is salt formation preferred over column chromatography for this intermediate?
A: Salt formation converts the intermediate into a crystalline solid, allowing for purification via simple filtration and recrystallization. This eliminates the need for expensive and solvent-intensive column chromatography, significantly reducing processing time and cost while improving scalability.
Q: How does this method address palladium catalyst residue issues?
A: The process involves filtering off the solid Pd/C catalyst immediately after hydrogenation. Subsequent salt formation and recrystallization steps further exclude metal impurities, ensuring the final intermediate meets stringent heavy metal specifications required for API synthesis.
Q: What acids are suitable for the salt formation step?
A: The patent specifies that various inorganic and organic acids can be used, including hydrochloric acid, phosphoric acid, and maleic acid. Hydrochloric acid is particularly preferred for its effectiveness in yielding high-purity solids with excellent recovery rates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable PARP Inhibitor Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent literature to commercial reality requires deep technical expertise and robust manufacturing infrastructure. We have extensively evaluated the salt formation strategy disclosed in CN111278830B and possess the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facilities are equipped with state-of-the-art hydrogenation reactors and crystallization units capable of executing this precise chemistry under stringent purity specifications. We understand that the quality of the intermediate dictates the success of the final API, which is why our rigorous QC labs employ advanced analytical techniques to verify that every batch meets the high purity standards (e.g., >99.5% HPLC) demonstrated in the patent examples. Our commitment to quality ensures that you receive a material that is ready for immediate coupling, minimizing your internal processing burden.
We invite global pharmaceutical partners to collaborate with us on optimizing their supply chains for PARP inhibitors. By leveraging our expertise in this novel purification technology, we can help you achieve significant cost reduction in pharmaceutical intermediates manufacturing while securing a stable supply of critical materials. We encourage you to contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. Our team is ready to provide specific COA data and route feasibility assessments to demonstrate how we can add value to your project. Let us handle the complexities of synthesis so you can focus on bringing life-saving therapies to patients faster.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →