Scalable Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles for Advanced Pharmaceutical Intermediates
Scalable Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles for Advanced Pharmaceutical Intermediates
The pharmaceutical and fine chemical industries are constantly seeking robust, cost-effective pathways to access nitrogen-containing heterocycles, particularly those bearing trifluoromethyl groups which are known to enhance metabolic stability and lipophilicity in drug candidates. Patent CN110467579B discloses a groundbreaking preparation method for 5-trifluoromethyl substituted 1,2,4-triazole compounds, addressing critical bottlenecks in current synthetic methodologies. This technology leverages a non-metallic iodine-promoted cyclization strategy that bypasses the need for expensive transition metal catalysts or harsh reaction conditions typically associated with trifluoromethylation. For R&D directors and procurement specialists, this represents a significant opportunity to streamline the supply chain for high-value pharmaceutical intermediates. The method utilizes cheap and readily available starting materials, specifically hydrazones and trifluoroethylimidoyl chlorides, ensuring that the production of these vital scaffolds can be achieved with substantial cost savings and operational simplicity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of trifluoromethylated nitrogen heterocycles has been fraught with challenges that hinder efficient commercial manufacturing. Traditional approaches often rely on the direct trifluoromethylation of pre-synthesized heterocyclic cores, a process that necessitates the use of specialized and often hazardous trifluoromethylating reagents which drive up raw material costs significantly. Alternatively, existing methods may employ trifluoroethylimide acid halides but frequently suffer from limited substrate scope or require rigorous anhydrous and anaerobic conditions that complicate reactor operations and increase energy consumption. Furthermore, many established protocols depend on heavy metal catalysts, introducing severe complications regarding residual metal limits in final API intermediates and creating burdensome waste disposal requirements. These factors collectively result in prolonged lead times and inflated production budgets, making it difficult for supply chain managers to maintain consistent availability of high-purity building blocks.
The Novel Approach
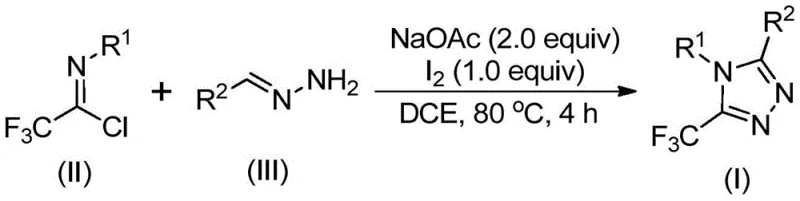
In stark contrast, the methodology described in patent CN110467579B introduces a streamlined, iodine-promoted synthesis that fundamentally alters the economic and operational landscape of triazole production. By reacting trifluoroethylimidoyl chloride with hydrazones in the presence of sodium acetate and elemental iodine, the process achieves high conversion rates under relatively mild thermal conditions (80-100°C). This novel route eliminates the dependency on precious metal catalysts, thereby removing the need for complex metal scavenging steps during purification. The reaction proceeds efficiently in common organic solvents like dichloroethane (DCE) without the strict requirement for inert atmospheres, drastically simplifying the engineering controls needed for commercial scale-up of complex pharmaceutical intermediates. This shift not only enhances safety profiles but also opens the door for more flexible manufacturing schedules and reduced overhead costs.
Mechanistic Insights into Iodine-Promoted Cyclization
From a mechanistic perspective, this transformation is a sophisticated example of base-promoted oxidative cyclization that ensures high regioselectivity and yield. The reaction likely initiates with a base-mediated intermolecular carbon-nitrogen bond formation between the hydrazone and the trifluoroethylimidoyl chloride, generating a trifluoroacetamidine intermediate. Subsequent isomerization sets the stage for the critical oxidative step, where elemental iodine acts as a mild oxidant to facilitate iodination. This is followed by an intramolecular electrophilic substitution and aromatization sequence that locks in the 1,2,4-triazole core with the trifluoromethyl group precisely positioned at the 5-position. Understanding this mechanism is vital for process chemists aiming to optimize high-purity pharmaceutical intermediates, as it highlights the dual role of iodine as both a promoter and an oxidant, avoiding the generation of toxic byproducts associated with stronger oxidizing agents.
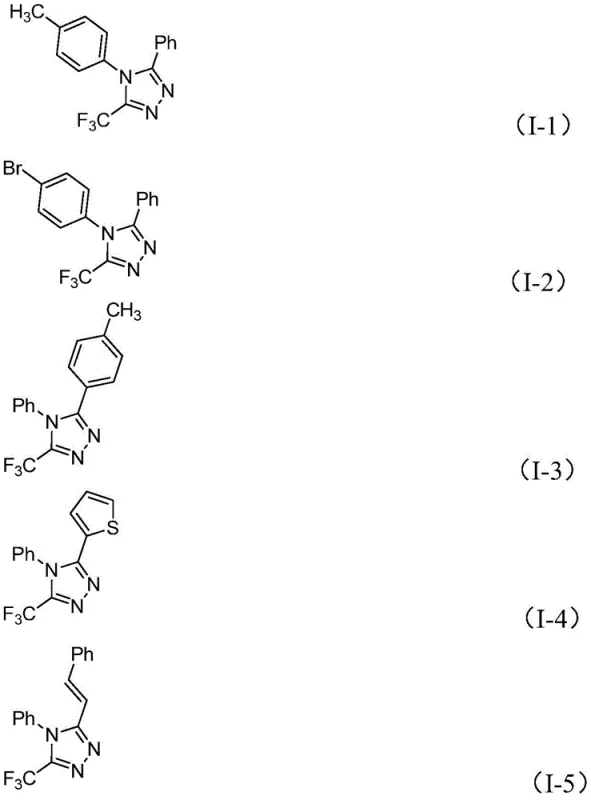
Furthermore, the substrate tolerance of this reaction is exceptionally broad, allowing for the incorporation of diverse electronic and steric environments without compromising yield. As demonstrated in the patent examples, various substituted aryl groups—including electron-donating methyl and methoxy groups as well as electron-withdrawing bromo and nitro functionalities—are well-tolerated at both the R1 and R2 positions. Even heteroaryl systems like thiophene and furan, as well as alkenyl chains, can be successfully integrated into the triazole scaffold. This versatility is crucial for medicinal chemists who require rapid access to diverse analog libraries for structure-activity relationship (SAR) studies. The ability to synthesize compounds like I-1 through I-5 with yields ranging from moderate to excellent underscores the robustness of this catalytic system in handling complex molecular architectures.
How to Synthesize 5-Trifluoromethyl-1,2,4-Triazoles Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires adherence to specific stoichiometric ratios and thermal profiles to maximize efficiency. The process begins by combining sodium acetate, trifluoroethylimidoyl chloride, and the chosen hydrazone substrate in a suitable organic solvent, with dichloroethane being the preferred medium for optimal solubility and reaction kinetics. The detailed standardized synthesis steps, including precise molar equivalents and workup procedures, are outlined in the guide below to ensure reproducibility and safety during operation.
- Combine sodium acetate, trifluoroethylimidoyl chloride, and hydrazone substrates in an organic solvent such as dichloroethane.
- Heat the reaction mixture to 80-100°C and maintain stirring for 2 to 4 hours to facilitate initial condensation.
- Introduce elemental iodine to the system, continue heating for 1 to 2 hours, then perform standard filtration and chromatographic purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this iodine-promoted methodology offers tangible strategic benefits that extend beyond mere chemical novelty. The primary advantage lies in the drastic simplification of the raw material portfolio; by utilizing commodity chemicals like hydrazones and simple imidoyl chlorides instead of exotic fluorinating agents, companies can secure a more stable and resilient supply chain. This reduction in material complexity directly translates to cost reduction in pharmaceutical intermediate manufacturing, as the volatility and high price points associated with specialized trifluoromethyl reagents are completely avoided. Additionally, the elimination of heavy metal catalysts removes a significant regulatory hurdle, streamlining the quality control process and reducing the time required for batch release.
- Cost Reduction in Manufacturing: The economic impact of this process is profound due to the substitution of expensive catalytic systems with inexpensive elemental iodine and sodium acetate. By removing the necessity for palladium, copper, or other transition metals, manufacturers save significantly on catalyst procurement costs and, more importantly, on the downstream processing required to remove metal residues to ppm levels. This leaner material input profile allows for a more competitive pricing structure for the final triazole products, enhancing margin potential for downstream API production.
- Enhanced Supply Chain Reliability: The reliance on widely available starting materials such as aromatic amines and aldehydes (precursors to hydrazones) ensures that production is not bottlenecked by the scarcity of niche reagents. Since these precursors are produced on a massive global scale for various industries, the risk of supply disruption is minimized. This reliability is critical for maintaining continuous manufacturing operations and meeting the stringent delivery timelines demanded by global pharmaceutical clients, effectively reducing lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The operational simplicity of running reactions at 80-100°C without strict inert gas protection makes this process inherently scalable from gram to multi-kilogram batches. The absence of toxic heavy metals and the use of manageable solvents simplify waste stream treatment, aligning with increasingly strict environmental regulations. This ease of scale-up facilitates the rapid transition from process development to commercial production, ensuring that new drug candidates can be supported with sufficient material volumes without requiring extensive process re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this 5-trifluoromethyl-1,2,4-triazole synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent literature, providing a clear picture of the method's capabilities and limitations for potential adopters.
Q: What are the primary advantages of this iodine-promoted method over traditional trifluoromethylation?
A: Unlike conventional methods that often require expensive trifluoromethylating reagents or toxic heavy metal catalysts, this protocol utilizes inexpensive elemental iodine and readily available hydrazones, significantly lowering raw material costs and simplifying waste treatment.
Q: Is this synthetic route suitable for large-scale industrial production?
A: Yes, the process is highly scalable as it operates under mild conditions (80-100°C) without strict anhydrous or anaerobic requirements, and the workup involves simple filtration and column chromatography, making it ideal for commercial scale-up.
Q: What is the substrate scope for the R1 and R2 groups in this triazole synthesis?
A: The method exhibits excellent functional group tolerance, successfully accommodating various substituted aryl groups (including methyl, methoxy, bromo, and nitro substituents) as well as heteroaryl and alkenyl groups at the R1 and R2 positions.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that efficient heterocycle synthesis plays in accelerating drug discovery and development. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the rigorous demands of global pharmaceutical partners. We are committed to delivering products with stringent purity specifications, supported by our rigorous QC labs that utilize advanced analytical techniques to verify every batch. Our capability to adapt innovative technologies like the iodine-promoted cyclization described in CN110467579B allows us to offer superior value propositions to our clients.
We invite you to engage with our technical procurement team to discuss how this advanced synthesis route can be tailored to your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits of switching to this metal-free methodology. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, ensuring that your supply chain is optimized for both performance and profitability.