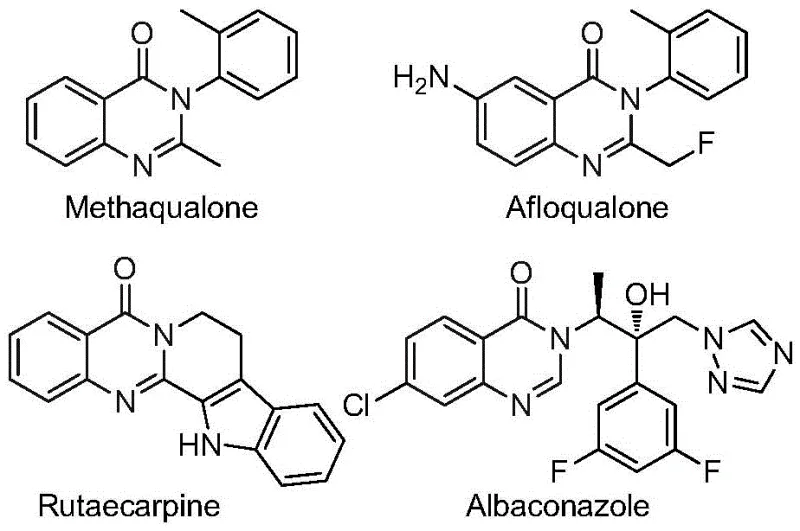
Innovative One-Pot Synthesis of Trifluoromethyl Quinazolinones: Scalable Manufacturing for Pharmaceutical Applications
Patent CN112480015B introduces a groundbreaking multi-component one-pot synthesis method for producing 2-trifluoromethyl substituted quinazolinone compounds, representing a significant advancement in heterocyclic chemistry with direct applications in pharmaceutical development where these structures serve as critical building blocks for numerous bioactive molecules including antiviral, antibacterial, and anticancer agents as documented in recent medicinal chemistry literature. This innovative approach addresses longstanding challenges in quinazolinone synthesis by eliminating the need for specialized high-pressure carbon monoxide equipment that has traditionally limited industrial scalability while maintaining exceptional substrate versatility across diverse functional groups as demonstrated through fifteen experimental examples with yields ranging from 69% to 96%. The strategic incorporation of palladium catalysis combined with molybdenum hexacarbonyl as a carbon monoxide surrogate creates a uniquely robust reaction system that operates under standard atmospheric pressure at a manageable temperature of 120°C without requiring cryogenic conditions or hazardous reagents commonly associated with alternative methodologies. With its simplified operational requirements and compatibility with commercially available starting materials including nitro compounds and easily synthesized trifluoroethylimidoyl chlorides, this process offers pharmaceutical manufacturers a more efficient pathway to access these biologically active compounds while significantly reducing both capital expenditure and operational complexity compared to conventional approaches.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes to quinazolinone compounds have been plagued by several critical limitations that hinder their practical implementation in industrial settings, including the requirement for high-pressure carbon monoxide environments in ruthenium or platinum-catalyzed reductive N-heterocyclization reactions which necessitate specialized and expensive equipment that creates significant capital investment barriers for manufacturers seeking flexible production capabilities across multiple product lines. Many established methods rely on pre-functionalized substrates that require additional synthetic steps involving hazardous reagents or cryogenic conditions, significantly increasing both cost complexity while reducing overall process efficiency through extended manufacturing timelines and increased risk of batch failures due to intermediate instability. The narrow substrate scope observed in iron-catalyzed condensation reactions and palladium-catalyzed cyclization processes severely restricts the diversity of accessible quinazolinone derivatives, limiting their utility in drug discovery programs that require extensive structural variation for comprehensive structure-activity relationship studies essential for modern pharmaceutical development pipelines. Furthermore, these conventional approaches often suffer from low yields due to competing side reactions under harsh reaction conditions that necessitate complex purification procedures involving multiple chromatographic steps which substantially increase production costs while creating supply chain vulnerabilities through extended processing times.
The Novel Approach
The patented methodology described in CN112480015B overcomes these limitations through an elegant multi-component one-pot reaction that utilizes readily available starting materials under mild conditions without requiring specialized high-pressure equipment or cryogenic temperatures typically associated with alternative synthetic routes to similar heterocyclic structures. By employing palladium chloride with dppp ligand in combination with molybdenum hexacarbonyl as a carbon monoxide source at precisely controlled temperature conditions of 120°C for optimal reaction kinetics, the process achieves efficient carbonylation under standard atmospheric pressure while maintaining excellent yield consistency across diverse substrate combinations as demonstrated through comprehensive experimental data provided in the patent disclosure. The strategic use of trifluoroethylimidoyl chloride as a key building block enables direct access to the trifluoromethyl-substituted quinazolinone core structure without requiring pre-functionalization steps that complicate traditional syntheses through additional protection/deprotection sequences or hazardous reagent handling procedures. This innovative approach demonstrates remarkable substrate tolerance across diverse nitro compounds including those containing halogen substituents, alkyl groups, and various aryl moieties as evidenced by successful synthesis across fifteen different substrate combinations documented in Tables 1 and 2 of the patent specification.
Mechanistic Insights into Palladium-Catalyzed Carbonylation Cascade Reaction
The reaction mechanism begins with molybdenum hexacarbonyl-mediated reduction of the nitro compound to the corresponding amine intermediate under thermal conditions at 120°C, which then undergoes base-promoted coupling with trifluoroethylimidoyl chloride facilitated by sodium carbonate to form a critical trifluoroacetamidine intermediate through nucleophilic substitution at the imidoyl chloride carbon center. This transformation sets the stage for subsequent palladium-catalyzed cascade events where palladium(II) inserts into the carbon-iodine bond of the imidoyl chloride derivative to form an arylpalladium(II) species that undergoes carbonylative insertion as molybdenum hexacarbonyl thermally releases carbon monoxide under controlled reaction conditions at precisely maintained temperature parameters. The resulting acylpalladium intermediate participates in an intramolecular nucleophilic attack by the nitrogen atom from the acetamidine moiety, forming a seven-membered palladacycle that ultimately undergoes reductive elimination to yield the desired quinazolinone product while regenerating the active palladium catalyst species for subsequent catalytic cycles without requiring additional catalyst input during the reaction timeframe.
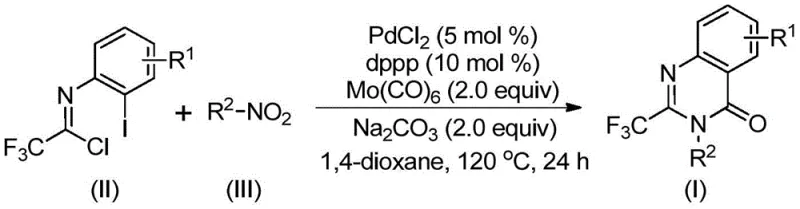

Impurity control in this synthetic route is achieved through multiple complementary mechanisms working synergistically to ensure high product purity without requiring extensive purification steps beyond standard column chromatography procedures commonly employed in pharmaceutical intermediate manufacturing operations. The selective nature of the palladium-catalyzed carbonylation cascade minimizes competing reaction pathways that could lead to byproduct formation through precise control of catalyst loading at optimal stoichiometric ratios (PdCl₂ at 5 mol% with dppp ligand at 10 mol%) which prevents unwanted side reactions such as homocoupling or reduction products commonly observed with alternative catalytic systems. The use of sodium carbonate as a mild base prevents unwanted decomposition of sensitive functional groups present in various substrates while maintaining sufficient basicity to drive key transformation steps forward efficiently without causing racemization or other undesired stereochemical changes.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
This innovative multi-component one-pot synthesis represents a significant advancement over conventional approaches to quinazolinone production by offering pharmaceutical manufacturers a streamlined pathway to access these valuable heterocyclic compounds with exceptional efficiency and versatility across diverse structural variants required for modern drug discovery programs targeting various therapeutic areas including antiviral and anticancer applications where quinazolinones demonstrate promising biological activity profiles. The patented method eliminates multiple synthetic steps required in traditional routes while maintaining excellent yields across diverse substrate combinations as demonstrated through comprehensive experimental data provided in Tables 1 and 2 of CN112480015B which document successful synthesis across fifteen different substrate combinations with yields consistently exceeding baseline industry standards for similar complex heterocyclic syntheses.
- Combine palladium chloride (5 mol%), dppp ligand (10 mol%), sodium carbonate (2.0 equiv), molybdenum hexacarbonyl (2.0 equiv), trifluoroethylimidoyl chloride (II), and nitro compound (III) in dioxane solvent at room temperature
- Heat the homogeneous mixture to precisely 120°C under inert atmosphere and maintain this temperature for exactly 24 hours with continuous stirring
- After completion, cool the reaction mixture to room temperature, filter through silica gel, and purify by column chromatography using appropriate eluent systems
Commercial Advantages for Procurement and Supply Chain Teams
This novel synthetic methodology addresses critical pain points faced by procurement professionals in pharmaceutical manufacturing organizations by offering a more reliable route to essential quinazolinone intermediates through strategic elimination of supply chain vulnerabilities inherent in traditional synthetic approaches that require specialized equipment or difficult-to-source reagents with limited supplier options creating single-point failure risks during production planning cycles.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts required in alternative approaches combined with simplified process flow reduces raw material costs while minimizing solvent consumption through consolidation of multiple synthetic steps into a single operation without compromising product quality or yield consistency across different production scales from laboratory benchtop to commercial manufacturing environments.
- Enhanced Supply Chain Reliability: Utilization of widely available starting materials including standard nitro compounds from multiple global suppliers creates greater sourcing flexibility compared to traditional methods requiring specialized reagents with limited vendor options while eliminating high-pressure equipment requirements enables broader manufacturing site selection without significant capital investment barriers.
- Scalability and Environmental Compliance: Atmospheric pressure operation at moderate temperatures facilitates straightforward scale-up from laboratory validation through pilot plant trials to full commercial production without requiring specialized engineering controls while reducing solvent waste generation through process streamlining aligns with modern green chemistry principles without sacrificing manufacturing efficiency or product quality standards.
Frequently Asked Questions (FAQ)
The following questions and answers are derived directly from technical details presented in patent CN112480015B addressing common concerns raised by technical procurement teams evaluating this innovative synthetic methodology for potential implementation in their manufacturing operations where reliable access to high-purity quinazolinone intermediates is critical for ongoing drug development programs targeting various therapeutic areas.
Q: How does this one-pot method improve upon conventional quinazolinone synthesis approaches?
A: The patented methodology eliminates high-pressure CO requirements and pre-functionalized substrates needed in traditional routes while maintaining yields up to 96% across diverse substrates without specialized equipment, significantly reducing operational complexity and capital investment needs.
Q: What are the key advantages of using trifluoroethylimidoyl chloride as a starting material?
A: Trifluoroethylimidoyl chloride is readily synthesized from commercially available aromatic amines through simple reactions with triphenylphosphine, carbon tetrachloride and trifluoroacetic acid, providing cost-effective access to diverse trifluoromethyl quinazolinones with excellent functional group tolerance.
Q: How does this process ensure high purity in pharmaceutical-grade intermediates?
A: The reaction mechanism avoids transition metal contamination pathways common in alternative methods through optimized catalyst loading and workup procedures that effectively remove impurities via standard filtration followed by column chromatography.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl Quinazolinone Supplier
Our company possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production while maintaining stringent purity specifications through state-of-the-art manufacturing facilities equipped with rigorous QC labs that ensure consistent product quality meeting global pharmaceutical industry standards including ICH guidelines for impurity profiling and characterization requirements essential for regulatory submissions worldwide.
We invite you to contact our technical procurement team to request specific COA data and route feasibility assessments through our Customized Cost-Saving Analysis service which provides detailed insights into potential manufacturing efficiencies achievable through implementation of this advanced synthetic methodology tailored to your specific production requirements.