Scalable Metal-Free Synthesis of Substituted Indolizine Derivatives for Pharmaceutical Applications
Scalable Metal-Free Synthesis of Substituted Indolizine Derivatives for Pharmaceutical Applications
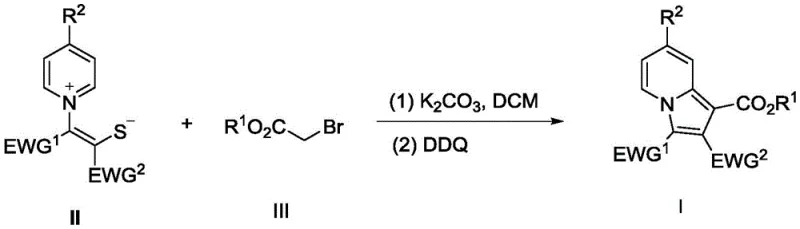
The rapid development of nitrogen-containing heterocyclic compounds remains a cornerstone of modern medicinal chemistry, particularly for scaffolds exhibiting significant biological activity such as anti-mitotic and cardiovascular properties. A groundbreaking approach detailed in patent CN111440165A introduces a highly efficient, metal-free methodology for constructing the indolizine core, a privileged structure found in numerous bioactive alkaloids and drug candidates. This innovation utilizes a sulfur-containing ylide and an α-bromocarbonyl compound as key starting materials, employing potassium carbonate as a base and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as a mild oxidant. By conducting the reaction in dichloromethane at ambient temperature without the need for inert gas protection, this process addresses critical bottlenecks in traditional heterocycle synthesis. For R&D directors and process chemists, this represents a paradigm shift towards greener, more cost-effective manufacturing of complex pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the indolizine skeleton has heavily relied on transition metal-catalyzed processes, such as copper-catalyzed [3+2] cycloadditions or oxidative linkages involving expensive palladium or rhodium complexes. While these methods possess undeniable academic utility, they present severe limitations for industrial scale-up, primarily due to the high loading of costly metal catalysts and the necessity for excess oxidants. The presence of residual heavy metals in the final product necessitates complex and expensive purification protocols to comply with stringent ICH Q3D guidelines for elemental impurities in drug substances. Furthermore, these traditional routes often require harsh reaction conditions, including elevated temperatures and strict inert atmospheres, which increase energy consumption and operational complexity. The structural rigidity of some catalytic systems also limits substrate scope, preventing the efficient synthesis of diverse derivatives required for structure-activity relationship (SAR) studies.
The Novel Approach
In stark contrast, the methodology disclosed in the referenced patent offers a streamlined, one-pot solution that circumvents the need for any transition metal catalysts. By leveraging the nucleophilicity of sulfur-containing ylides and the electrophilicity of α-bromocarbonyl compounds, the reaction initiates a cascade that builds the fused ring system under remarkably mild conditions. The use of inorganic bases like potassium carbonate and common organic oxidants like DDQ allows the transformation to proceed smoothly at room temperature (25°C). This elimination of precious metals not only drastically reduces raw material costs but also simplifies the workup procedure, as there is no need for specialized metal scavengers or extensive washing steps. The robustness of this metal-free protocol ensures high yields and excellent purity profiles, making it an ideal candidate for the commercial scale-up of complex pharmaceutical intermediates where cost and quality are paramount.
Mechanistic Insights into Metal-Free Oxidative Cyclization
The core of this synthetic breakthrough lies in the strategic interaction between the sulfur ylide and the activated halide, followed by an oxidative aromatization step. Initially, the sulfur ylide acts as a 1,3-dipole equivalent, attacking the α-bromocarbonyl compound to form a transient intermediate. The presence of the base facilitates the deprotonation and subsequent intramolecular cyclization, effectively closing the five-membered ring onto the pyridine nitrogen. This step is critical as it establishes the indolizine framework without requiring external thermal energy to overcome high activation barriers. Following the cyclization, the addition of DDQ serves as the driving force for the final aromatization, removing two hydrogen atoms to establish the fully conjugated aromatic system characteristic of the indolizine core.
This mechanistic pathway offers superior control over impurity profiles compared to radical-based metal catalysis. Because the reaction proceeds through defined ionic intermediates rather than uncontrolled radical species, the formation of side products such as homocoupled dimers or over-oxidized byproducts is minimized. The mild nature of the oxidant (DDQ) ensures that sensitive functional groups on the substrate, such as esters or ethers, remain intact during the process. For quality control teams, this translates to a cleaner crude reaction mixture, which significantly reduces the burden on downstream purification units. The ability to tune the electronic properties of both the ylide and the bromide component allows for precise modulation of the reaction kinetics, ensuring consistent reproducibility across different batches of high-purity indolizine derivatives.
How to Synthesize Substituted Indolizine Derivatives Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires adherence to specific stoichiometric ratios and sequential addition protocols to maximize yield and minimize waste. The process is designed to be operationally simple, utilizing standard glassware and common solvents, which lowers the barrier for adoption in diverse manufacturing facilities. The key to success lies in monitoring the consumption of the starting ylide before introducing the oxidant, ensuring that the cyclization is complete prior to the aromatization step. Detailed standardized operating procedures for this transformation are outlined below to guide process engineers in replicating these results.
- Dissolve the sulfur-containing ylide (Compound II), alpha-bromocarbonyl compound (Compound III), and potassium carbonate in dichloromethane solvent at room temperature.
- Monitor the reaction until Compound II is completely consumed, then add 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as the oxidant to promote aromatization.
- Upon completion, remove the organic solvent under reduced pressure and purify the crude residue using silica gel column chromatography with petroleum ether and ethyl acetate.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this metal-free methodology offers transformative advantages that directly impact the bottom line and operational resilience. By removing the dependency on volatile transition metal markets and expensive ligand systems, manufacturers can achieve substantial cost savings in raw material acquisition. The simplified workflow reduces the number of unit operations required, leading to shorter cycle times and increased throughput capacity without the need for capital-intensive equipment upgrades. Furthermore, the use of readily available, commodity-grade reagents enhances supply chain security, mitigating the risks associated with sourcing specialized catalytic systems from single-source vendors.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts removes a significant cost driver associated with both the purchase of precious metals and the disposal of metal-contaminated waste streams. Without the need for expensive metal scavengers or complex extraction protocols to meet regulatory limits, the overall cost of goods sold (COGS) is significantly reduced. Additionally, the ability to run the reaction at room temperature eliminates energy costs associated with heating or cooling reactors, further enhancing the economic viability of large-scale production runs.
- Enhanced Supply Chain Reliability: The starting materials, including sulfur ylides and alpha-bromocarbonyls, are commercially available from multiple global suppliers, ensuring a robust and competitive supply chain. This diversity of supply sources prevents bottlenecks and allows procurement managers to negotiate better terms. The stability of the reagents also means they can be stored for extended periods without degradation, allowing for strategic stockpiling to buffer against market fluctuations or logistical disruptions.
- Scalability and Environmental Compliance: The one-pot nature of the reaction minimizes solvent usage and waste generation, aligning with green chemistry principles and reducing environmental compliance costs. The absence of heavy metals simplifies wastewater treatment and solid waste disposal, lowering the environmental footprint of the manufacturing process. This eco-friendly profile is increasingly valuable for pharmaceutical companies aiming to meet sustainability goals and regulatory standards for green manufacturing practices.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel indolizine synthesis route. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity for technical teams evaluating this technology for integration into their existing pipelines.
Q: Why is the metal-free nature of this indolizine synthesis critical for API manufacturing?
A: Traditional methods often rely on expensive transition metal catalysts like copper, which require rigorous and costly removal steps to meet strict heavy metal limits in pharmaceutical ingredients. This novel protocol eliminates transition metals entirely, significantly simplifying downstream purification and reducing the risk of metal contamination in the final active pharmaceutical ingredient.
Q: What are the specific reaction conditions required for this oxidative cyclization?
A: The process operates under exceptionally mild conditions, specifically at room temperature (25°C) in dichloromethane. Unlike many heterocyclic syntheses that demand inert gas protection or high heat, this method proceeds efficiently in air without specialized equipment, making it highly suitable for large-scale industrial production.
Q: Does this synthetic route support a wide variety of substituent groups?
A: Yes, the method demonstrates broad substrate tolerance. It accommodates various electron-withdrawing groups such as methoxycarbonyl, ethoxycarbonyl, and benzoyl groups, as well as different substituents on the pyridine ring, allowing for the diverse functionalization required in medicinal chemistry campaigns.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Substituted Indolizine Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting innovative synthetic routes that balance efficiency, purity, and cost-effectiveness. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from lab-scale discovery to full-scale manufacturing is seamless. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of substituted indolizine derivatives meets the highest industry standards for pharmaceutical applications.
We invite you to collaborate with our technical team to explore how this metal-free synthesis can optimize your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the potential economic benefits tailored to your volume needs. We encourage you to contact our technical procurement team today to obtain specific COA data and comprehensive route feasibility assessments, ensuring your supply chain is built on a foundation of reliability and scientific excellence.