Advanced Aminolysis Strategy for High-Purity Lafutidine Intermediates and Commercial Scale-Up
Advanced Aminolysis Strategy for High-Purity Lafutidine Intermediates and Commercial Scale-Up
The global demand for high-quality gastrointestinal therapeutics continues to drive innovation in the synthesis of key active pharmaceutical ingredients (APIs) and their precursors. Lafutidine, a potent and long-acting histamine H2 receptor antagonist, represents a critical molecule in this sector, offering superior gastric mucosal protection compared to earlier generation drugs. However, the commercial viability of Lafutidine has historically been constrained by complex purification challenges associated with its synthetic route. A pivotal breakthrough in this domain is documented in patent CN102212060A, which introduces a novel aminolysis-based methodology. This technical insight report analyzes the transformative potential of this patent, specifically focusing on how replacing traditional hydrazinolysis with amine-mediated deprotection fundamentally alters the impurity profile and economic feasibility of Lafutidine production. By leveraging this advanced chemistry, manufacturers can achieve significantly higher purity standards while streamlining the supply chain for this valuable pharmaceutical intermediate.
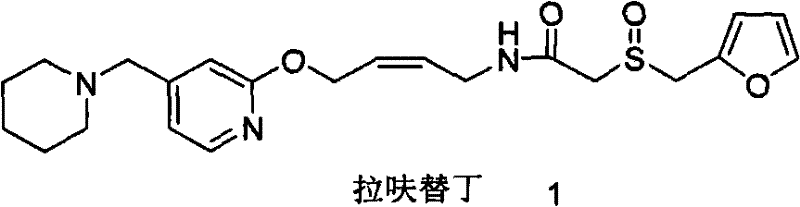
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of Lafutidine has relied heavily on the Gabriel synthesis method, a classic approach for preparing primary amines. This conventional route involves the formation of an N-substituted phthalimide followed by decomposition using hydrazine hydrate, known as the Ing-Manske reaction. While chemically sound in theory, this method presents severe practical drawbacks when applied to the sensitive Lafutidine scaffold. The primary issue lies in the use of hydrazine hydrate, which acts not only as a nucleophile for deprotection but also as a reducing agent. Under the reflux conditions necessary for the reaction, hydrazine inadvertently reduces the unsaturated double bond in the butenyl chain of the molecule. This side reaction generates a persistent impurity known as dihydro lafutidine, which possesses physicochemical properties nearly identical to the target API.
Furthermore, the removal of this dihydro impurity is notoriously difficult. Standard purification techniques such as repeated recrystallization or column chromatography often fail to reduce the impurity content below stringent regulatory thresholds without catastrophic losses in yield. Literature data indicates that crude products from hydrazinolysis typically contain 1.5-2.0% of this reduced impurity. Even after aggressive recrystallization processes designed to lower this content to acceptable levels (around 0.5%), the overall yield of the synthetic route plummets to approximately 45-50%. This inefficiency creates a bottleneck for reliable API intermediate supplier operations, driving up costs and complicating waste management due to the extensive solvent usage required for purification.
The Novel Approach
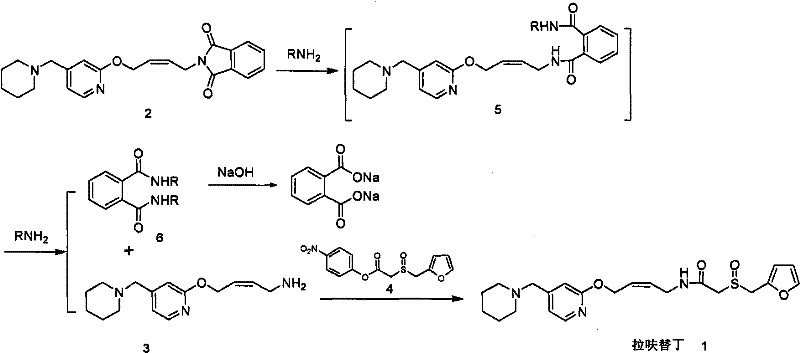
In stark contrast to the hydrazine-dependent legacy methods, the technology disclosed in CN102212060A utilizes a direct aminolysis strategy that circumvents the formation of reduction byproducts entirely. This innovative approach employs simple amines, such as methylamine, ethylamine, or ammonia, to cleave the phthalimide protecting group. By substituting the reducing hydrazine with non-reducing amines, the integrity of the alkene double bond in the Lafutidine backbone is preserved throughout the deprotection step. The process proceeds through a distinct intermediate, identified as an N-alkyl-phthalic diamide (Formula 5), which subsequently reacts further to release the desired primary amine (Formula 3). This mechanistic shift ensures that the crude product is virtually free of the dihydro lafutidine impurity from the outset.
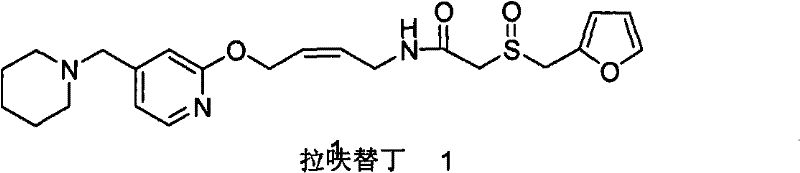
The operational advantages of this novel approach are substantial for cost reduction in pharmaceutical manufacturing. Because the crude product generated via aminolysis lacks the stubborn dihydro impurity, the need for multiple, yield-destructive recrystallization steps is eliminated. The patent data highlights that this method can achieve a total recovery of 85-95% from the starting phthalimide maleate, nearly doubling the efficiency of the traditional route. Additionally, the byproducts formed during aminolysis, such as soluble phthalic acid derivatives, are easily separated from the organic phase through simple aqueous workups. This simplification of the downstream processing not only enhances throughput but also aligns with green chemistry principles by reducing solvent consumption and waste generation, making it an ideal candidate for commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Amine-Mediated Phthalimide Cleavage
To fully appreciate the robustness of this synthesis, one must examine the mechanistic details of the aminolysis reaction. The process begins with the nucleophilic attack of the amine (RNH2) on one of the carbonyl carbons of the phthalimide ring in Formula 2. This attack opens the imide ring, resulting in the formation of the intermediate Formula 5, which is an N-alkyl-phthalic diamide. Unlike the hydrazinolysis pathway, which involves the formation of a cyclic phthalazine-1,4-dione byproduct that drives the reaction forward but introduces reducing equivalents, the aminolysis pathway generates a linear diamide intermediate. This intermediate is stable enough to be isolated if desired, as demonstrated in the patent examples where methylamine reaction yields a solid precipitate of Formula 5. However, for process efficiency, the reaction is typically continued in the same pot.
Upon prolonged exposure to the amine or subsequent treatment with a base like sodium hydroxide, the second amide bond in Formula 5 is cleaved. This step releases the free primary amine (Formula 3) and converts the phthalic moiety into a water-soluble salt, such as disodium phthalate. This solubility difference is the key to the high purity achieved. The dihydro lafutidine impurity, which plagues the hydrazine method, is structurally formed via the reduction of the C=C double bond. Since amines like methylamine or ethylamine lack the reducing power of hydrazine under these mild conditions (typically 5-50°C), the alkene remains intact. Consequently, the final condensation of Formula 3 with the activated ester (Formula 4) yields Lafutidine with a purity profile that inherently meets strict ICH guidelines without requiring extensive remediation.
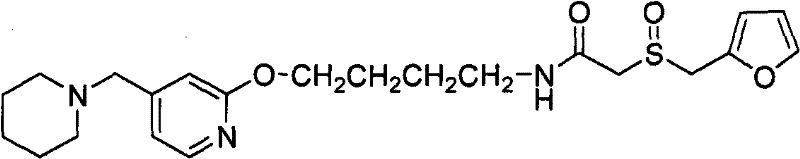
How to Synthesize Lafutidine Efficiently
The implementation of this aminolysis protocol offers a straightforward path for process chemists aiming to optimize Lafutidine production. The method is versatile regarding the amine source, accommodating ammonia, methylamine, ethylamine, or propylamine, with methylamine and ethylamine aqueous solutions being particularly effective. The reaction can be conducted in various solvents including alcohols, ethers, or mixtures with water, providing flexibility for existing manufacturing infrastructure. The following guide outlines the standardized synthesis steps derived from the patent data, ensuring high yield and purity.
- React N-(4-(4-piperidino methyl) pyridyl-2-oxo)-2-alkene butyl phthalimide (Formula 2) with an amine such as methylamine or ethylamine to generate the intermediate phthalic diamide (Formula 5).
- Continue the reaction of Formula 5 with the amine to fully cleave the phthalimide group, yielding the primary amine intermediate (Formula 3) and a soluble phthalic acid derivative byproduct.
- Condense the purified Formula 3 amine with furfuryl sulfinyl acetate p-nitrophenyl phenolic ester (Formula 4) to obtain high-purity Lafutidine without dihydro impurities.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition from hydrazinolysis to aminolysis represents a strategic opportunity to enhance margin and reliability. The traditional reliance on hydrazine hydrate introduces significant safety and handling costs, as hydrazine is toxic, potentially carcinogenic, and requires specialized containment and disposal protocols. By switching to common alkylamines, the facility reduces its hazardous material footprint, thereby lowering compliance costs and insurance premiums associated with dangerous goods storage. Furthermore, the dramatic improvement in yield—from roughly 50% to over 85%—directly translates to a reduction in the cost of goods sold (COGS). This efficiency gain means that less raw material is required to produce the same amount of finished API, effectively insulating the supply chain from volatility in the pricing of upstream precursors like the phthalimide maleate.
- Cost Reduction in Manufacturing: The elimination of multiple recrystallization steps is the primary driver of cost savings in this new process. In the conventional method, recovering yield losses from purification often requires processing double the volume of material to meet output targets. The aminolysis method produces a crude product of such high quality that minimal purification is needed. This reduction in unit operations decreases energy consumption, labor hours, and solvent procurement costs. Additionally, the avoidance of hydrazine removes the need for expensive quenching and detoxification procedures, further streamlining the operational expenditure profile for large-scale production runs.
- Enhanced Supply Chain Reliability: Sourcing high-purity intermediates is often a bottleneck in pharmaceutical supply chains. The aminolysis route utilizes commodity chemicals (methylamine, ethylamine, NaOH) that are readily available in bulk quantities globally, unlike specialized reagents that might have limited suppliers. This abundance ensures that production schedules are not disrupted by reagent shortages. Moreover, the robustness of the reaction conditions (0-150°C, preferably 5-50°C) allows for flexible manufacturing across different climates and facility types. The ability to consistently deliver material with <0.1% critical impurities strengthens the trust between the supplier and the downstream API manufacturer, reducing the risk of batch rejections and audit findings.
- Scalability and Environmental Compliance: Scaling chemical processes often amplifies waste issues, but this aminolysis method is inherently cleaner. The byproducts are water-soluble salts that can be easily separated and treated in standard wastewater facilities, unlike the complex organic waste streams generated by hydrazine reactions. The high atom economy and reduced solvent usage align with modern environmental, social, and governance (ESG) goals. For a reliable agrochemical intermediate supplier or pharma partner, demonstrating a greener synthesis route is increasingly becoming a prerequisite for vendor qualification. This process facilitates the commercial scale-up of complex pharmaceutical intermediates by minimizing the engineering controls needed for hazardous reagents and maximizing the throughput per reactor volume.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the aminolysis synthesis of Lafutidine. These insights are derived directly from the experimental data and comparative examples provided in the patent literature, offering clarity on impurity profiles, yield expectations, and intermediate handling. Understanding these nuances is essential for technical teams evaluating the feasibility of adopting this superior manufacturing route.
Q: Why does the traditional hydrazinolysis method produce dihydro lafutidine impurities?
A: The traditional method uses hydrazine hydrate, which acts as a reducing agent under reflux conditions. This causes the hydrogenation reduction of the unsaturated double bond in the butenyl chain, forming dihydro lafutidine, a structurally similar impurity that is difficult to remove via recrystallization.
Q: What are the yield advantages of the aminolysis method described in CN102212060A?
A: The aminolysis method achieves a total recovery of 85-95% from the Formula 2 maleate starting material. In contrast, the conventional hydrazinolysis route typically yields only 45-50% after the multiple recrystallization steps required to lower impurity levels.
Q: Can the intermediate Formula 5 be isolated during the process?
A: Yes, the intermediate suitable-N-alkyl-N-(4-(4-piperidino methyl) pyridyl-2-oxo)-2-alkene butyl phthalic diamide (Formula 5) can be isolated as a solid, particularly when using methylamine. However, for industrial efficiency, the process allows for a 'one-pot' continuation to Formula 3 without isolation.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Lafutidine Supplier
The technological advancements detailed in patent CN102212060A underscore the importance of partnering with a manufacturer that possesses deep chemical expertise and the capacity for innovation. NINGBO INNO PHARMCHEM stands at the forefront of this evolution, leveraging our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the precise temperature controls and solvent systems required for the aminolysis process, ensuring that every batch of Lafutidine intermediate meets stringent purity specifications. With our rigorous QC labs and commitment to continuous process improvement, we guarantee a supply of high-purity pharmaceutical intermediates that empower your drug development pipelines.
We invite global partners to collaborate with us to unlock the full economic potential of this optimized synthesis. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to discuss specific COA data, route feasibility assessments, and how our advanced aminolysis capabilities can secure your supply chain for the long term. Let us be your trusted partner in delivering high-quality Lafutidine intermediates to the global market.