Scalable Synthesis of N-Difluoromethyl Azaindoles for Advanced Pharmaceutical Applications
Scalable Synthesis of N-Difluoromethyl Azaindoles for Advanced Pharmaceutical Applications
The introduction of fluorine-containing groups, particularly the difluoromethyl (CF2H) moiety, has become a cornerstone strategy in modern medicinal chemistry for optimizing the pharmacokinetic profiles of drug candidates. As detailed in the recent patent disclosure CN112279849A, a novel and highly efficient synthetic methodology has been developed for the preparation of N-difluoromethyl azaindole compounds. This technology addresses critical bottlenecks in the synthesis of these valuable heterocyclic scaffolds, which serve as essential building blocks for a wide array of bioactive molecules including kinase inhibitors and antiviral agents. The disclosed process leverages a metal-free catalytic system that operates under remarkably mild conditions, typically at room temperature, thereby eliminating the need for energy-intensive heating protocols often associated with traditional difluoromethylation reactions. By utilizing readily available and cost-effective reagents such as ethyl bromodifluoroacetate, this innovation offers a robust pathway for generating high-purity intermediates with exceptional functional group tolerance.
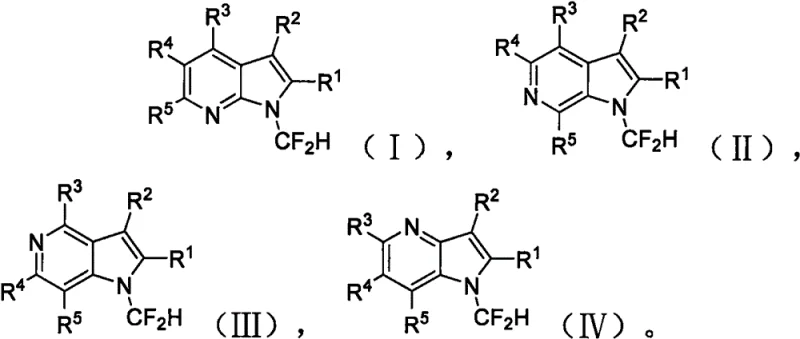
The structural versatility of the resulting N-difluoromethyl azaindoles is extensive, accommodating a broad spectrum of substituents at multiple positions on the pyrrolopyridine core. As illustrated in the general formula, the method supports substrates bearing electron-withdrawing groups like nitro and cyano, as well as electron-donating groups such as methyl and alkoxy, without compromising yield or selectivity. This adaptability is crucial for pharmaceutical research and development teams who require rapid access to diverse analog libraries for structure-activity relationship (SAR) studies. Furthermore, the absence of transition metal catalysts in this protocol significantly simplifies the downstream purification process, ensuring that the final products meet the stringent purity specifications required for clinical trial materials. For procurement managers and supply chain directors, this represents a tangible opportunity to secure a reliable pharmaceutical intermediate supplier capable of delivering complex fluorinated scaffolds with reduced lead times and lower overall manufacturing costs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the installation of difluoromethyl groups onto nitrogen-containing heterocycles has been plagued by significant technical and economic challenges that hinder industrial scalability. Early methodologies, such as those reported by the Hu Jinbo group, relied heavily on chlorodifluoromethylbenzenesulfone as the difluoromethylating agent, a reagent that is not only prohibitively expensive but also poses handling difficulties in large-scale operations.
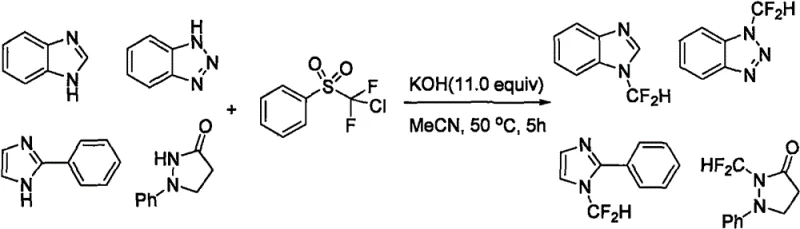
As depicted in the reaction scheme above, these traditional routes often necessitate the use of strong bases like potassium hydroxide in acetonitrile at elevated temperatures, which can lead to decomposition of sensitive functional groups on the substrate. Other approaches, such as the method developed by the Shouggchang group using triphenylphosphonium difluoroacetate inner salts, suffer from similar economic drawbacks due to the high cost of the phosphonium reagent. Additionally, protocols reported by Prakash involving trifluoromethyltrimethylsilane require extreme reaction temperatures reaching 170°C in triglyme, creating severe safety hazards and energy consumption issues that are incompatible with green chemistry principles. These legacy methods frequently result in complex impurity profiles that demand rigorous and costly purification steps, such as preparative HPLC, to remove trace metal contaminants or phosphine oxides, thereby inflating the cost of goods sold (COGS) for the final active pharmaceutical ingredient.
The Novel Approach
In stark contrast to these cumbersome legacy processes, the technology described in patent CN112279849A introduces a streamlined, metal-free difluoromethylation strategy that fundamentally reshapes the economic landscape of azaindole synthesis. This novel approach utilizes ethyl bromodifluoroacetate (BrCF2CO2Et) as the primary difluorocarbene source, a reagent that is commercially abundant, significantly cheaper, and environmentally friendlier than its sulfone or phosphonium counterparts. The reaction proceeds efficiently in common organic solvents such as acetonitrile, dichloromethane, or ethyl acetate, utilizing mild inorganic or organic bases like potassium tert-butoxide or cesium carbonate. Crucially, the reaction can be conducted at room temperature for unsubstituted azaindoles, or with mild heating up to 80°C for more sterically hindered substrates, drastically reducing the energy footprint of the manufacturing process. This shift from harsh, high-energy conditions to ambient temperature processing not only enhances operational safety but also preserves the integrity of labile functional groups, allowing for the direct synthesis of complex intermediates without the need for extensive protecting group strategies.
Mechanistic Insights into Base-Mediated Difluorocarbene Generation
The mechanistic elegance of this synthesis lies in the in situ generation of difluorocarbene species through a base-mediated elimination pathway, which subsequently undergoes nucleophilic attack by the azaindole nitrogen. Upon treatment with a suitable base such as potassium tert-butoxide, ethyl bromodifluoroacetate undergoes alpha-elimination to release carbon dioxide and ethanol, generating the highly reactive difluorocarbene (:CF2) intermediate. This electrophilic carbene is then rapidly trapped by the nucleophilic nitrogen atom of the azaindole ring system, forming the stable N-CF2H bond. The choice of base is critical; strong non-nucleophilic bases facilitate the rapid generation of the carbene species while minimizing side reactions such as hydrolysis of the ester moiety. Experimental data indicates that potassium tert-butoxide provides optimal results, likely due to its solubility profile in acetonitrile and its ability to effectively deprotonate the intermediate without promoting degradation of the sensitive azaindole core. The reaction kinetics are favorable, typically reaching completion within 6 to 12 hours, which suggests a low activation energy barrier for the carbene transfer step under these specific solvent conditions.
From an impurity control perspective, the metal-free nature of this mechanism offers distinct advantages for regulatory compliance and product quality. Traditional transition-metal catalyzed cross-couplings often leave behind trace amounts of palladium, copper, or nickel, which are strictly regulated in final drug substances and require specialized scavenging resins to remove. In this base-mediated protocol, the only byproducts are inorganic salts (e.g., potassium bromide) and volatile organic compounds (ethanol, CO2), which are easily removed during the aqueous workup and concentration steps. This clean reaction profile minimizes the formation of difficult-to-remove organometallic impurities, thereby simplifying the purification workflow to standard silica gel column chromatography or recrystallization. For R&D directors, this translates to a more predictable impurity profile and a faster timeline for method validation and technology transfer to pilot plants, ensuring that the supply of high-purity pharmaceutical intermediates remains uninterrupted and compliant with ICH Q3D guidelines for elemental impurities.
How to Synthesize N-Difluoromethyl Azaindole Efficiently
The practical implementation of this synthesis route is designed for ease of operation, requiring standard laboratory equipment and avoiding the need for specialized high-pressure reactors or inert gas gloveboxes beyond standard nitrogen line techniques. The process begins with the dissolution of the azaindole substrate and the difluoromethylating reagent in a polar aprotic solvent, followed by the controlled addition of the base to initiate carbene generation. Detailed standard operating procedures regarding stoichiometry, addition rates, and quenching protocols are essential for maximizing yield and reproducibility across different batch sizes.
- Dissolve the azaindole substrate, difluorocarbene reagent (e.g., BrCF2CO2Et), and alkali base in an organic solvent like acetonitrile.
- Stir the reaction mixture at room temperature or mild heating (up to 80°C) for 6 to 24 hours under nitrogen atmosphere.
- Quench with water, extract with ethyl acetate, dry over sodium sulfate, and purify the crude product via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis method offers compelling strategic advantages that directly impact the bottom line and operational resilience. The primary driver of value is the substantial cost reduction in API manufacturing achieved by replacing exotic, high-cost reagents with commodity chemicals. Ethyl bromodifluoroacetate is produced on a multi-ton scale globally, ensuring a stable supply chain that is not subject to the volatility often seen with specialized fluorinating agents. Furthermore, the elimination of expensive transition metal catalysts removes the need for costly metal scavenging steps and the associated validation testing, leading to significant savings in both material costs and analytical overhead.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven by the use of low-cost raw materials and the avoidance of energy-intensive heating cycles. By operating at room temperature or mild heating, the facility utility costs for steam and cooling are drastically minimized compared to processes requiring reflux at 170°C. Additionally, the simplified workup procedure, which relies on basic extraction and filtration rather than complex chromatographic separations for metal removal, reduces solvent consumption and waste disposal costs. This lean manufacturing approach allows for a more competitive pricing structure for the final N-difluoromethyl azaindole intermediates, providing a clear margin advantage for downstream drug development projects.
- Enhanced Supply Chain Reliability: Supply chain continuity is significantly bolstered by the reliance on widely available reagents that are not sourced from single-supplier monopolies. The robustness of the reaction conditions means that production is less susceptible to disruptions caused by equipment failures or variations in raw material quality. The high functional group tolerance ensures that a single standardized protocol can be applied to a wide variety of azaindole derivatives, reducing the need for multiple specialized production lines and inventory SKUs. This flexibility enables suppliers to respond rapidly to changing demand signals from pharmaceutical clients, reducing lead time for high-purity pharmaceutical intermediates and ensuring just-in-time delivery for critical clinical trial materials.
- Scalability and Environmental Compliance: The environmental profile of this synthesis aligns perfectly with modern green chemistry mandates, facilitating easier regulatory approval and community acceptance for manufacturing sites. The absence of heavy metals eliminates the risk of soil and water contamination, while the generation of benign byproducts like CO2 and ethanol simplifies wastewater treatment requirements. From a scalability standpoint, the exothermic nature of the carbene generation is manageable under standard jacketed reactor conditions, allowing for safe scale-up from kilogram to multi-ton production without the need for specialized high-pressure containment. This inherent safety and environmental compatibility future-proofs the supply chain against tightening environmental regulations, ensuring long-term viability for commercial scale-up of complex pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this N-difluoromethylation technology. These insights are derived directly from the experimental data and comparative analysis presented in the patent literature, providing a factual basis for decision-making.
Q: What are the advantages of this N-difluoromethylation method over traditional protocols?
A: Unlike traditional methods requiring expensive reagents like chlorodifluoromethylbenzenesulfone or harsh temperatures up to 170°C, this protocol utilizes cheap ethyl bromodifluoroacetate at room temperature without transition metals.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the method features mild reaction conditions, easily available raw materials, and simple purification steps, making it highly adaptable for commercial scale-up of complex pharmaceutical intermediates.
Q: What is the substrate scope for this difluoromethylation reaction?
A: The method demonstrates excellent functional group tolerance, successfully modifying various azaindole derivatives substituted with methyl, nitro, chloro, bromo, cyano, and ester groups.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable N-Difluoromethyl Azaindole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced fluorination technologies play in accelerating the discovery and development of next-generation therapeutics. Our team of expert process chemists has thoroughly evaluated the methodology described in CN112279849A and integrated its core principles into our proprietary manufacturing platforms. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition seamlessly from benchtop discovery to full-scale commercial supply. Our state-of-the-art facilities are equipped with rigorous QC labs and stringent purity specifications to guarantee that every batch of N-difluoromethyl azaindole meets the highest international standards for pharmaceutical applications.
We invite you to collaborate with us to leverage this cutting-edge synthesis route for your specific drug development programs. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your target molecule, demonstrating exactly how this metal-free protocol can optimize your budget. We encourage you to contact us today to discuss your requirements,索取 specific COA data for our existing library of azaindole derivatives, and obtain comprehensive route feasibility assessments for your custom synthesis projects. Let us be your trusted partner in delivering high-quality fluorinated intermediates that drive your innovation forward.