Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial API Production
Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial API Production
The pharmaceutical industry continuously seeks robust synthetic methodologies to access nitrogen-containing heterocycles, particularly those functionalized with trifluoromethyl groups, due to their profound impact on drug metabolism and bioavailability. Patent CN111423381B discloses a groundbreaking preparation method for 2-trifluoromethyl substituted imidazole compounds, addressing critical challenges in modern medicinal chemistry. This technology leverages a transition metal palladium-catalyzed carbonylation series reaction, utilizing readily available starting materials such as trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salts. The significance of this innovation lies in its ability to construct complex molecular architectures under exceptionally mild conditions, specifically at 30°C, which contrasts sharply with traditional high-energy synthetic routes. As a reliable pharmaceutical intermediate supplier, understanding such advancements is crucial for developing next-generation therapeutics where metabolic stability and lipophilicity are paramount design criteria.
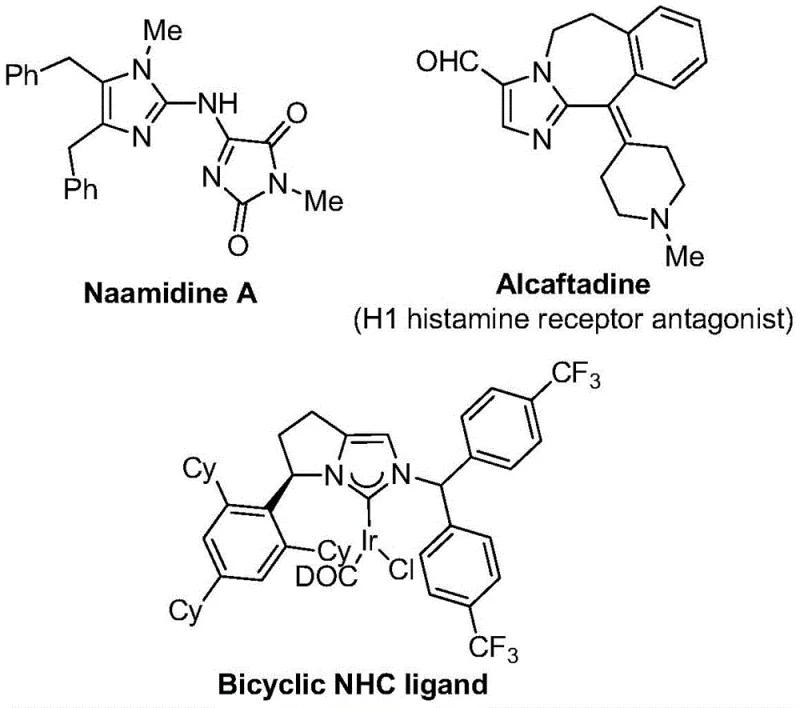
The structural versatility of imidazole derivatives is evident in their widespread presence in bioactive natural products and synthetic drugs, as illustrated by compounds like Naamidine A and the H1 histamine receptor antagonist Alcaftadine. The introduction of a trifluoromethyl group at the 2-position of the imidazole ring further enhances these properties by significantly improving electronegativity and metabolic resistance. However, accessing these specific motifs has historically been hindered by the reliance on hazardous or expensive trifluoromethyl synthons. The disclosed method overcomes these barriers by employing a modular approach that allows for the precise installation of diverse aryl groups at the 1 and 5 positions of the imidazole core. This flexibility is essential for medicinal chemists aiming to optimize structure-activity relationships (SAR) during the lead optimization phase of drug discovery, ensuring that potential candidates possess the necessary physicochemical profiles for clinical success.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoromethyl-substituted nitrogen-containing heterocycles has relied heavily on direct reactions between substrates and specialized trifluoromethyl synthons. Common reagents in this domain include trifluorodiazoethane and various trifluoroethylimidoyl halides, which, while effective, present significant logistical and safety challenges for large-scale manufacturing. Trifluorodiazoethane, for instance, is known for its instability and potential explosiveness, requiring specialized handling equipment and stringent safety protocols that drive up operational costs. Furthermore, many conventional methods necessitate harsh reaction conditions, such as elevated temperatures or strong acidic/basic environments, which can lead to the degradation of sensitive functional groups often present in complex drug intermediates. These limitations restrict the substrate scope, making it difficult to synthesize diversified libraries of compounds required for comprehensive biological screening. Additionally, the use of stoichiometric amounts of expensive reagents and the generation of substantial chemical waste contribute to a less sustainable and economically viable process, creating bottlenecks for supply chain continuity in the production of high-purity pharmaceutical intermediates.
The Novel Approach
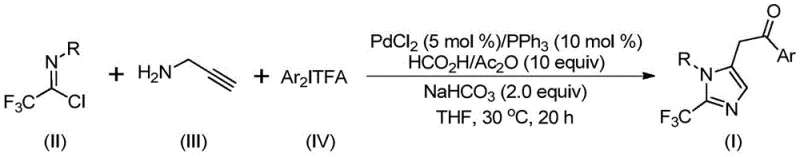
In stark contrast to these legacy techniques, the novel approach detailed in the patent utilizes a palladium-catalyzed cascade reaction that operates under remarkably mild and controlled conditions. By employing trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salts as the primary building blocks, the method achieves high reaction efficiency with excellent atom economy. The core of this transformation involves a sophisticated intermolecular carbon-nitrogen bond formation promoted by alkali, followed by isomerization and palladation steps that construct the imidazole ring skeleton. Crucially, the carbonyl group required for the final structure is introduced safely using a formic acid/acetic anhydride mixture as a carbon monoxide surrogate, eliminating the need for handling toxic CO gas directly. This strategy not only enhances operational safety but also broadens the functional group tolerance, allowing for the incorporation of electron-donating and electron-withdrawing groups such as methoxy, nitro, and halogens without compromising yield. The result is a streamlined, scalable process that delivers 2-trifluoromethyl imidazoles with high purity, making it an ideal candidate for cost reduction in API manufacturing.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
The mechanistic pathway of this transformation is a testament to the elegance of modern organometallic catalysis, involving a sequence of well-defined elementary steps that ensure high selectivity. The reaction initiates with the formation of a trifluoroacetamidine intermediate through an intermolecular carbon-nitrogen bond formation between the trifluoroethylimidoyl chloride and propargylamine, facilitated by the base. Following this, a critical isomerization occurs, setting the stage for the palladium cycle. The palladium catalyst, generated in situ from PdCl2 and triphenylphosphine, undergoes oxidative addition or coordination with the alkyne moiety, leading to a palladation event that forms an alkenyl palladium intermediate. This species subsequently isomerizes to a more stable alkyl palladium intermediate, positioning the metal center for the subsequent carbonylation step. The carbon monoxide, released slowly from the formic acid/acetic anhydride additive, inserts into the palladium-carbon bond to generate an acyl palladium intermediate. This step is pivotal as it installs the ketone functionality adjacent to the forming ring system.
The final stages of the catalytic cycle involve the activation of the diaryl iodonium salt. This hypervalent iodine reagent undergoes oxidative addition to the palladium center, generating a high-valent tetravalent palladium intermediate. This transient species is highly reactive and rapidly undergoes reductive elimination to forge the final carbon-carbon bond, releasing the desired 2-trifluoromethyl-substituted imidazole product and regenerating the active palladium catalyst. This intricate dance of oxidation states and ligand exchanges is carefully balanced by the reaction conditions, specifically the use of THF as a solvent and sodium bicarbonate as a mild base. The choice of these reagents minimizes side reactions such as homocoupling of the alkyne or decomposition of the sensitive imidoyl chloride. Furthermore, the mechanism inherently suppresses the formation of regioisomers, ensuring that the trifluoromethyl group remains strictly at the 2-position of the imidazole ring. This level of control is vital for regulatory compliance, as impurity profiles in pharmaceutical intermediates must be tightly managed to meet stringent quality standards required by global health authorities.
How to Synthesize 2-Trifluoromethyl Imidazole Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires strict adherence to the optimized parameters outlined in the patent to maximize yield and purity. The process is designed to be operationally simple, avoiding the need for specialized high-pressure equipment or cryogenic conditions. The key to success lies in the precise stoichiometry of the reagents, particularly the ratio of the palladium catalyst to the ligand and the base. The protocol suggests a molar ratio of palladium chloride to triphenylphosphine to sodium bicarbonate of approximately 0.05:0.1:2, ensuring sufficient catalytic turnover while maintaining a basic environment to neutralize acidic byproducts. The reaction time is another critical variable, with 16 to 24 hours at 30°C identified as the optimal window to drive the reaction to completion without degrading the product. For detailed procedural instructions, including specific workup and purification techniques, please refer to the standardized guide below.
- Combine palladium chloride, triphenylphosphine, sodium bicarbonate, acetic anhydride, formic acid, trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salt in an organic solvent such as THF.
- Stir the reaction mixture at a mild temperature of 30°C for a duration of 16 to 24 hours to ensure complete conversion.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target 2-trifluoromethyl substituted imidazole compound.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented methodology offers transformative benefits for procurement managers and supply chain directors looking to optimize their sourcing strategies for heterocyclic intermediates. The shift towards this new synthetic route addresses several pain points associated with traditional manufacturing, primarily focusing on cost efficiency, supply reliability, and environmental sustainability. By utilizing commodity chemicals like propargylamine and commercially available diaryl iodonium salts, the dependency on custom-synthesized, high-cost specialty reagents is significantly reduced. This democratization of raw materials means that supply chains are less vulnerable to disruptions caused by the limited availability of niche precursors. Moreover, the mild reaction conditions translate directly into lower energy consumption, as there is no need for prolonged heating or cooling cycles, contributing to a smaller carbon footprint and reduced utility costs for the manufacturing facility.
- Cost Reduction in Manufacturing: The economic advantages of this process are driven by the use of inexpensive catalysts and reagents combined with a simplified workflow. Unlike methods requiring expensive noble metals or complex ligand systems, this protocol utilizes palladium chloride and triphenylphosphine, which are cost-effective and widely accessible. The elimination of hazardous gases like carbon monoxide from the supply chain removes the need for costly safety infrastructure and monitoring systems. Furthermore, the high conversion rates and clean reaction profiles minimize the burden on downstream purification processes, reducing the consumption of silica gel and solvents during column chromatography. These factors collectively contribute to a substantial reduction in the overall cost of goods sold (COGS), allowing for more competitive pricing in the global market for pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the robustness and flexibility of the synthetic route. The method's tolerance for a wide array of functional groups means that a single platform technology can be used to produce a diverse library of analogues, reducing the need for multiple distinct manufacturing lines. The starting materials, including various substituted aromatic amines and aryl boronic acids (precursors to the iodonium salts), are commodity chemicals with established global supply networks. This ensures consistent availability and mitigates the risk of shortages that often plague the production of complex fine chemicals. Additionally, the scalability of the reaction from gram to kilogram levels without significant loss in efficiency provides confidence for long-term planning and capacity allocation, ensuring that production targets can be met reliably even during periods of high demand.
- Scalability and Environmental Compliance: Environmental, Health, and Safety (EHS) compliance is increasingly a deciding factor in vendor selection, and this process excels in this regard. The use of formic acid and acetic anhydride as a CO source is a greener alternative to using pressurized carbon monoxide cylinders, significantly lowering the risk profile of the operation. The reaction generates minimal hazardous waste, and the solvents used, such as THF, can be readily recovered and recycled, aligning with green chemistry principles. The simplicity of the post-treatment procedure, involving filtration and standard chromatography, reduces the volume of aqueous waste streams requiring treatment. These attributes make the process highly scalable, facilitating the transition from R&D to commercial production with fewer regulatory hurdles. For companies aiming to reduce their environmental impact while maintaining high output, adopting this methodology represents a strategic advantage in meeting corporate sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this synthesis method, derived directly from the experimental data and specifications provided in the patent documentation. Understanding these nuances is essential for technical teams evaluating the feasibility of integrating this chemistry into their existing workflows. The answers reflect the specific conditions and outcomes observed during the development of the process, ensuring accuracy and relevance for practical application.
Q: What are the optimal reaction conditions for this palladium-catalyzed synthesis?
A: According to patent CN111423381B, the reaction proceeds efficiently at a mild temperature of 30°C using THF as the solvent. The catalyst system typically employs PdCl2 and PPh3 with sodium bicarbonate as a base, reacting for 16 to 24 hours.
Q: Can this method tolerate diverse functional groups on the aryl rings?
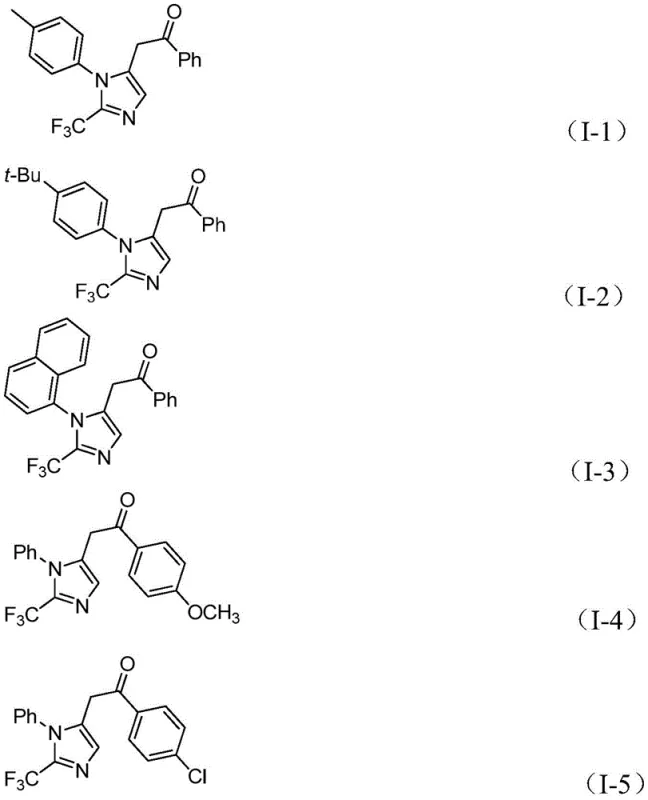
A: Yes, the method demonstrates excellent substrate compatibility. It successfully accommodates various substituents including methyl, tert-butyl, halogens (chlorine, bromine), trifluoromethyl, and nitro groups on both the imidoyl chloride and the diaryl iodonium salt components.
Q: What is the source of the carbonyl group in the final imidazole structure?
A: The carbonyl functionality is introduced via a carbonylation reaction where carbon monoxide is generated in situ from the decomposition of formic acid and acetic anhydride, acting as a safe and effective CO surrogate.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Imidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced synthetic methodologies play in accelerating drug discovery and development. Our team of expert chemists is well-versed in the intricacies of palladium-catalyzed transformations and is fully equipped to leverage the innovations described in patent CN111423381B for your specific project needs. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop to plant floor is seamless and efficient. Our state-of-the-art facilities are designed to handle complex organic syntheses with precision, adhering to stringent purity specifications and rigorous QC labs protocols to guarantee the highest quality intermediates. Whether you require custom synthesis of novel analogues or large-scale supply of established compounds, our commitment to technical excellence and operational integrity makes us the ideal partner for your pharmaceutical supply chain.
We invite you to collaborate with us to explore the full potential of this cutting-edge technology for your pipeline. By partnering with NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and target specifications. Our technical procurement team is ready to assist you in navigating the complexities of chemical sourcing, providing specific COA data and route feasibility assessments to ensure your project stays on track and within budget. Contact us today to discuss how we can support your journey from molecule to medicine with reliable, high-quality 2-trifluoromethyl imidazole intermediates.