Innovative Iodine-Catalyzed Synthesis of High-Purity Trifluoromethyl Triazoles for Scalable Pharmaceutical Manufacturing
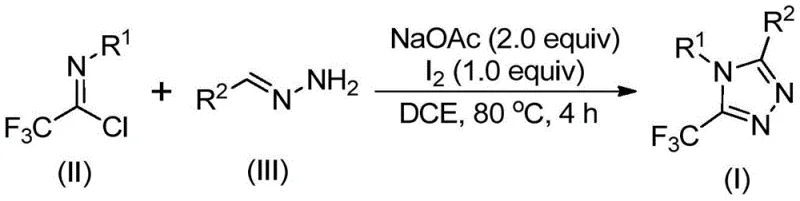
Patent CN110467579B presents a groundbreaking methodology for synthesizing 5-trifluoromethyl substituted 1,2,4-triazole compounds through an innovative iodine-catalyzed cyclization process that eliminates the need for stringent anhydrous and anaerobic conditions while avoiding toxic heavy metal catalysts entirely. This novel approach leverages readily available starting materials including sodium acetate, trifluoroethylimidoyl chloride, and hydrazone derivatives to construct the triazole core under mild thermal conditions at 80°C in dichloroethane solvent. The process demonstrates exceptional substrate flexibility by accommodating diverse aryl and heteroaryl substitutions at both R¹ and R² positions without requiring specialized handling procedures or expensive purification techniques. Crucially, this method achieves high yields across multiple derivatives while maintaining operational simplicity that facilitates seamless transition from laboratory-scale synthesis to industrial manufacturing environments. The elimination of transition metal catalysts not only reduces environmental impact but also circumvents complex metal removal steps that typically complicate pharmaceutical intermediate production workflows. This patent represents a significant advancement in heterocyclic chemistry that directly addresses critical pain points in the synthesis of fluorinated triazole compounds essential for modern drug discovery pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to synthesizing trifluoromethyl-substituted triazole compounds have been severely constrained by their dependence on either pre-formed heterocyclic scaffolds requiring subsequent trifluoromethylation or specialized trifluoromethyl-containing synthons that often demand rigorous anhydrous and anaerobic reaction environments to prevent decomposition or side reactions. These conventional methodologies frequently employ expensive transition metal catalysts such as palladium or copper complexes that necessitate elaborate removal protocols to meet pharmaceutical purity standards, significantly increasing production costs and complicating regulatory compliance for drug substance manufacturing. The limited functional group tolerance observed in many existing routes restricts structural diversity and hinders the synthesis of complex derivatives required for advanced medicinal chemistry applications. Furthermore, the requirement for specialized equipment to maintain oxygen-free conditions creates substantial barriers to scalability and increases capital expenditure for manufacturing facilities seeking to produce these critical intermediates at commercial volumes. The inherent instability of certain trifluoromethylating reagents also contributes to inconsistent yields and batch-to-batch variability that undermine supply chain reliability for pharmaceutical companies dependent on consistent intermediate quality.
The Novel Approach
The patented methodology overcomes these longstanding challenges through a brilliantly simple yet highly effective iodine-mediated cyclization process that operates under ambient atmospheric conditions without requiring any special handling precautions or expensive inert gas systems. By utilizing elemental iodine as a non-toxic catalyst alongside inexpensive sodium acetate as a base promoter, this approach eliminates the need for costly transition metal catalysts while maintaining excellent reaction efficiency across a broad spectrum of substrate combinations. The process demonstrates remarkable functional group tolerance that accommodates various substituents including methyl, methoxy, bromo, nitro, and trifluoromethyl groups at multiple positions on both coupling partners. This unprecedented flexibility enables medicinal chemists to rapidly generate diverse compound libraries for structure-activity relationship studies without modifying the core synthetic protocol. The reaction's compatibility with standard laboratory glassware and straightforward workup procedure involving simple filtration followed by column chromatography significantly reduces operational complexity while ensuring high product purity suitable for pharmaceutical applications. Most notably, the method's scalability has been demonstrated from milligram to gram quantities with consistent yields, providing a clear pathway for industrial implementation without requiring substantial process re-engineering.
Mechanistic Insights into Iodine-Mediated Triazole Formation
The reaction mechanism proceeds through a well-defined sequence beginning with base-promoted intermolecular carbon-nitrogen bond formation between the trifluoroethylimidoyl chloride and hydrazone components to generate a key trifluoroacetamidine intermediate. This initial step occurs readily at elevated temperatures without requiring anhydrous conditions due to the stability of the imidoyl chloride functionality under the reaction environment. Subsequent isomerization of this intermediate facilitates the formation of a reactive species that undergoes base-promoted oxidative iodination by elemental iodine to create an iodinated intermediate crucial for the cyclization step. The iodine catalyst plays a dual role by both promoting the oxidation step and facilitating the final intramolecular electrophilic substitution that drives ring closure and aromatization to form the stable triazole product. This mechanistic pathway is particularly advantageous because it avoids the formation of persistent radical species or unstable intermediates that could lead to side product formation or reduced yields.
The strategic use of iodine as a catalyst provides exceptional control over impurity profiles by directing the reaction through a specific mechanistic pathway that minimizes competing side reactions commonly observed in alternative synthetic routes. The absence of transition metals eliminates potential contamination from heavy metal residues that would require extensive purification steps to remove below acceptable thresholds for pharmaceutical applications. Furthermore, the mild reaction conditions prevent decomposition of sensitive functional groups present in complex substrates while maintaining high regioselectivity for the desired triazole isomer formation. This precise control over reaction pathways translates directly into superior product purity with minimal byproduct formation that would otherwise require additional separation steps during manufacturing scale-up.
How to Synthesize Trifluoromethyl Triazoles Efficiently
This patented methodology represents a significant advancement in heterocyclic synthesis that addresses critical challenges in producing fluorinated triazole compounds essential for pharmaceutical development pipelines. The process demonstrates exceptional operational simplicity combined with remarkable substrate flexibility that enables medicinal chemists to rapidly access diverse compound libraries without modifying core synthetic parameters. By eliminating requirements for specialized equipment or hazardous reagents while maintaining high yields across multiple derivatives, this approach provides an ideal solution for both research-scale synthesis and commercial manufacturing applications. The following standardized procedure outlines the precise implementation steps required to consistently reproduce this innovative synthetic route with optimal results.
- Combine sodium acetate (2.0 equiv), trifluoroethylimidoyl chloride (II), and hydrazone (III) in dichloroethane solvent under ambient atmospheric conditions without requiring anhydrous or anaerobic setup.
- Heat the reaction mixture to precisely 80°C and maintain thermal control for exactly four hours to facilitate base-promoted carbon-nitrogen bond formation and intermediate isomerization.
- Introduce elemental iodine (1.0 equiv) into the reaction system and continue heating at constant temperature for one additional hour to complete oxidative iodination and final cyclization.
Commercial Advantages for Procurement and Supply Chain Teams
This innovative synthetic approach delivers substantial value across procurement and supply chain operations by addressing fundamental pain points associated with traditional manufacturing methods for fluorinated heterocyclic compounds. The elimination of specialized handling requirements and expensive catalyst systems creates immediate opportunities for cost optimization while enhancing production reliability through simplified process parameters that reduce vulnerability to supply chain disruptions.
- Cost Reduction in Manufacturing: The complete avoidance of transition metal catalysts eliminates both direct material costs associated with expensive palladium or copper complexes and substantial downstream processing expenses required for metal removal to meet pharmaceutical purity standards. This dual cost-saving mechanism significantly reduces overall production expenses while streamlining quality control procedures through simplified analytical requirements that no longer need monitoring heavy metal residues. The use of inexpensive starting materials including commercially available sodium acetate and readily synthesized hydrazone derivatives further contributes to economic efficiency without compromising product quality or consistency.
- Enhanced Supply Chain Reliability: The method's compatibility with standard manufacturing equipment and elimination of specialized anhydrous/anaerobic processing requirements substantially reduces vulnerability to supply chain disruptions by removing dependencies on specialized gas systems or moisture-sensitive reagents. The broad availability of starting materials from multiple global suppliers creates robust sourcing options that mitigate single-point failure risks while enabling flexible production scheduling based on regional material availability rather than process constraints.
- Scalability and Environmental Compliance: The straightforward reaction workup involving simple filtration followed by standard column chromatography enables seamless scale-up from laboratory to commercial production volumes without requiring specialized equipment modifications or complex process re-engineering. The absence of toxic heavy metals and reduced solvent usage compared to conventional methods significantly lowers environmental impact while meeting increasingly stringent regulatory requirements for sustainable chemical manufacturing practices.
Frequently Asked Questions (FAQ)
The following questions address common concerns regarding implementation of this patented technology based on detailed analysis of its technical specifications and commercial implications for pharmaceutical intermediate manufacturing operations.
Q: How does this method eliminate anhydrous/anaerobic requirements compared to conventional triazole synthesis?
A: The patented process utilizes stable imidoyl chloride intermediates that remain reactive under ambient atmospheric conditions without decomposition risks associated with traditional methods requiring moisture-sensitive reagents or oxygen-free environments.
Q: What cost advantages arise from avoiding heavy metal catalysts in this manufacturing process?
A: Eliminating transition metal catalysts removes both direct material costs and substantial downstream processing expenses related to metal removal procedures while simplifying quality control requirements for pharmaceutical purity standards.
Q: How does substrate design flexibility enable diverse trifluoromethyl triazole derivative synthesis?
A: The broad functional group tolerance accommodates various substituents including methyl, methoxy, bromo, nitro groups at multiple positions without modifying core reaction parameters or requiring specialized handling procedures.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl Triazole Supplier
Our company brings extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production while maintaining stringent purity specifications through rigorous QC labs equipped with state-of-the-art analytical instrumentation. This patented iodine-catalyzed methodology represents just one example of our commitment to developing innovative synthetic solutions that address critical challenges in pharmaceutical intermediate manufacturing while optimizing cost structures without compromising quality standards.
We invite you to request a Customized Cost-Saving Analysis from our technical procurement team to evaluate how this technology can be implemented in your specific manufacturing context along with access to detailed COA data and route feasibility assessments tailored to your production requirements.