Advanced Rhodium-Catalyzed Synthesis of Trifluoromethyl Dihydrophenanthrenes for Commercial Scale-Up
The pharmaceutical and fine chemical industries are constantly seeking efficient pathways to access complex scaffolds with high biological potential. Patent CN113511966B introduces a groundbreaking transition metal-catalyzed method for synthesizing trifluoromethyl-substituted dihydrophenanthrene compounds, a structural motif known for its significant anti-inflammatory and anticancer activities. This innovation addresses the critical need for robust synthetic methodologies that can deliver high-purity intermediates without the burden of extreme reaction conditions. By leveraging a specialized rhodium catalyst system, this technology enables the direct construction of the dihydrophenanthrene core through a [4+2] cyclization strategy, marking a substantial advancement over conventional multi-step approaches.
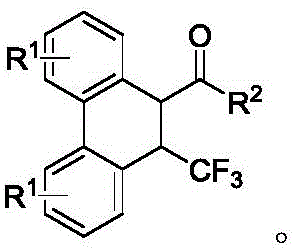
Dihydrophenanthrene derivatives are highly valued in medicinal chemistry due to their ability to inhibit inflammatory factors via pathways such as NF-κB. The introduction of a trifluoromethyl group further enhances the lipophilicity and metabolic stability of these molecules, often leading to improved drug-like properties. However, traditional synthetic routes to these compounds often suffer from low atom economy, harsh reaction conditions, or poor regioselectivity. The methodology described in this patent overcomes these limitations by utilizing 2-biphenylboronic acid compounds and α,β-unsaturated ketone compounds as readily accessible starting materials. The process operates under remarkably mild conditions, requiring only heating in an organic solvent in the presence of a rhodium catalyst and a silver salt oxidant, yet it achieves exceptional yields and selectivity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of polycyclic aromatic systems like dihydrophenanthrenes has relied on classical cyclization reactions that often demand stoichiometric amounts of strong acids or Lewis acids, leading to significant waste generation and safety hazards. Many existing protocols struggle with the precise installation of the trifluoromethyl group at the desired position, frequently resulting in complex mixtures of regioisomers that are difficult and costly to separate. Furthermore, traditional methods may require inert atmosphere protection and cryogenic temperatures, which drastically increase the operational complexity and energy consumption of the manufacturing process. These factors collectively contribute to higher production costs and longer lead times, creating bottlenecks for the reliable supply of high-purity pharmaceutical intermediates needed for drug development pipelines.
The Novel Approach
The novel approach detailed in CN113511966B represents a paradigm shift by employing a catalytic C-H activation strategy that is both atom-economical and operationally simple. The core of this innovation lies in the use of a rhodium catalyst featuring a bulky cyclopentadienyl ligand, specifically [CptBuRhI2]2. This specific ligand architecture plays a pivotal role in steering the reaction pathway towards the desired [4+2] cyclization product while suppressing competing 1,4-addition side reactions. The reaction proceeds efficiently in common organic solvents like ethyl acetate at moderate temperatures around 80°C. Crucially, the system is air-tolerant, eliminating the need for rigorous degassing or glovebox techniques. This simplicity translates directly into cost reduction in pharmaceutical intermediate manufacturing by reducing equipment requirements and processing time.
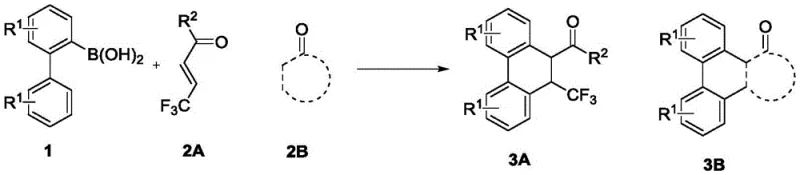
Mechanistic Insights into Rhodium-Catalyzed C-H Activation and Cyclization
Understanding the mechanistic underpinnings of this transformation is essential for R&D directors aiming to optimize the process for specific analogs. The reaction is initiated by the activation of the rhodium precursor, likely involving the silver oxidant to generate the active cationic rhodium species. This active catalyst then coordinates with the 2-biphenylboronic acid substrate, facilitating a directed C-H activation at the ortho-position of the biphenyl ring. The resulting rhodacycle intermediate is highly reactive and undergoes migratory insertion with the α,β-unsaturated ketone. The unique steric bulk of the CptBu ligand is critical at this stage; it creates a specific coordination environment that favors the formation of the six-membered ring through a second C-H activation or nucleophilic attack, followed by reductive elimination.
The patent provides compelling evidence for a two-step Michael addition mechanism, particularly noted in reactions involving benzoquinone derivatives where bridged ring compounds were isolated. This mechanistic clarity allows for precise impurity control, as the pathway to the major byproduct (the 1,4-addition product) is effectively blocked by the ligand design. The high selectivity ensures that the crude reaction mixture contains minimal impurities, simplifying downstream purification. For process chemists, this means that the method is not just a laboratory curiosity but a viable platform for generating diverse libraries of trifluoromethylated scaffolds with predictable outcomes, thereby accelerating the structure-activity relationship (SAR) studies required for lead optimization.
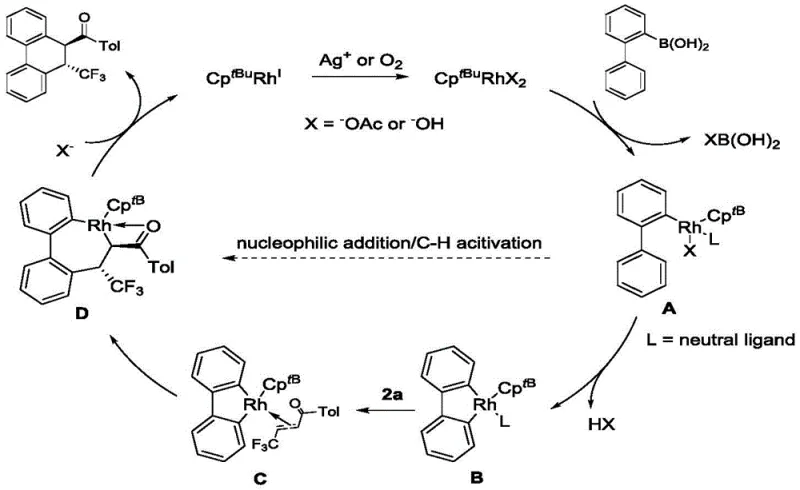
How to Synthesize Trifluoromethyl Substituted Dihydrophenanthrene Efficiently
The practical implementation of this synthesis is straightforward and designed for reproducibility. The general procedure involves mixing the 2-biphenylboronic acid derivative and the trifluoromethyl-substituted enone in a pressure-resistant tube. The catalyst [CptBuRhI2]2 is added alongside silver acetate and silver oxide as the oxidant system. Ethyl acetate serves as the preferred solvent due to its favorable safety profile and solubility characteristics. The reaction mixture is sealed and heated to 80°C, typically reaching completion within 12 hours as monitored by TLC. Upon cooling, standard aqueous workup and silica gel chromatography yield the pure product. The detailed standardized synthesis steps are provided in the guide below.
- Combine 2-biphenylboronic acid derivatives and alpha,beta-unsaturated ketone compounds in a reaction vessel with a rhodium catalyst such as [CptBuRhI2]2.
- Add silver salt oxidants like AgOAc and Ag2O along with an organic solvent such as ethyl acetate to the mixture.
- Heat the sealed reaction tube to 80°C under air conditions for approximately 12 hours, then purify the crude product via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this technology offers tangible strategic benefits beyond mere chemical novelty. The primary advantage lies in the significant simplification of the supply chain for raw materials. Both 2-biphenylboronic acids and α,β-unsaturated ketones are commodity chemicals available from multiple global suppliers, reducing the risk of single-source dependency. The robustness of the reaction conditions means that manufacturing can be performed in standard stainless steel reactors without the need for specialized lining or exotic metallurgy required for highly corrosive traditional methods. This compatibility with existing infrastructure facilitates a smoother technology transfer from lab to plant.
- Cost Reduction in Manufacturing: The elimination of expensive and hazardous reagents typically associated with classical cyclization methods leads to substantial cost savings. The high atom economy of the [4+2] cyclization ensures that a greater proportion of the starting material mass ends up in the final product, minimizing waste disposal costs. Furthermore, the air-tolerant nature of the reaction removes the capital expenditure associated with maintaining strict inert atmospheres, lowering the overall overhead for production facilities.
- Enhanced Supply Chain Reliability: The broad substrate scope demonstrated in the patent indicates that the process is resilient to variations in electronic and steric properties of the starting materials. This flexibility allows for the rapid sourcing of alternative substrates if supply disruptions occur. The high yields reported across numerous examples suggest a consistent and reliable process output, which is critical for maintaining continuous supply lines for downstream API synthesis.
- Scalability and Environmental Compliance: The patent explicitly includes data on scale-up experiments, confirming that the reaction efficiency is maintained at larger scales. This de-risks the commercialization pathway for new drug candidates. Additionally, the use of ethyl acetate, a greener solvent compared to chlorinated alternatives often used in similar transformations, aligns with increasingly stringent environmental regulations, reducing the regulatory burden and potential fines associated with volatile organic compound (VOC) emissions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding this synthesis method. These answers are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing clarity for stakeholders evaluating this technology for integration into their manufacturing portfolios.
Q: What are the key advantages of this rhodium-catalyzed method over traditional synthesis routes?
A: This method utilizes a bulky cyclopentadienyl ligand (CptBu) which effectively promotes the reductive elimination process prior to protonolysis, ensuring high selectivity for the [4+2] cyclized product rather than simple 1,4-addition byproducts. Additionally, the reaction proceeds under mild conditions (80°C) and is tolerant to air, simplifying operational requirements.
Q: Is this synthetic route suitable for large-scale manufacturing?
A: Yes, the patent demonstrates successful scale-up experiments where the reaction maintained high efficiency and yield even when the scale was increased significantly. The use of readily available starting materials and stable reaction conditions supports robust commercial scalability.
Q: What is the substrate scope for this trifluoromethylation reaction?
A: The method exhibits a broad substrate scope, accommodating various substituents on the biphenylboronic acid including alkyl, halogen, alkoxy, and trifluoromethyl groups. It also tolerates different aryl and heteroaryl groups on the enone partner, making it versatile for generating diverse chemical libraries.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl Dihydrophenanthrene Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced catalytic methodologies like the one described in CN113511966B for accelerating drug discovery and development. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop to manufacturing is seamless. We are committed to delivering high-purity pharmaceutical intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation.
We invite you to collaborate with us to leverage this cutting-edge synthesis technology for your specific project needs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how we can become your trusted partner in bringing next-generation therapeutics to market efficiently and reliably.