Advanced Palladium-Catalyzed Synthesis of Carbonyl-Bridged Biheterocyclic Compounds for Commercial Scale-Up
Advanced Palladium-Catalyzed Synthesis of Carbonyl-Bridged Biheterocyclic Compounds for Commercial Scale-Up
The pharmaceutical industry continuously seeks robust and scalable methodologies for constructing complex heterocyclic scaffolds, which serve as the core backbone for numerous bioactive molecules. Patent CN115353511A introduces a groundbreaking multi-component synthesis strategy for preparing carbonyl-bridged biheterocyclic compounds, specifically targeting the fusion of indolinone and imidazole motifs. This innovation addresses critical bottlenecks in traditional heterocycle synthesis by employing a transition metal palladium-catalyzed carbonylation cascade reaction. The significance of this technology lies in its ability to construct multiple chemical bonds in a single operational step, utilizing cheap and readily available starting materials such as trifluoroethylimidoyl chloride, propargylamine, and acrylamide derivatives. For R&D directors and process chemists, this represents a paradigm shift towards more efficient, atom-economical routes that minimize waste and maximize structural diversity. The method operates under remarkably mild conditions, typically at 30°C, which preserves sensitive functional groups and reduces energy consumption, making it an attractive candidate for the production of high-purity pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of biheterocyclic systems containing carbonyl bridges has been fraught with synthetic challenges that hinder efficient commercial production. Conventional literature reports primarily rely on three strategies: direct coupling of two pre-formed heterocyclic substrates, oxidative cyclization of substrates bearing dual nucleophiles, or multi-step sequences involving harsh conditions. The direct coupling approach often suffers from poor regioselectivity and requires expensive, pre-functionalized building blocks that drive up the cost of goods. Furthermore, traditional carbonylation reactions frequently necessitate the use of toxic carbon monoxide gas, which poses severe safety hazards and requires specialized high-pressure equipment and rigorous safety protocols, thereby increasing capital expenditure for manufacturing facilities. These legacy methods often lack the flexibility to accommodate diverse substituents, limiting the chemical space available for medicinal chemistry optimization. Additionally, multi-step syntheses inherently lead to lower overall yields due to cumulative losses at each isolation stage, generating significant amounts of solvent waste and increasing the environmental footprint of the process.
The Novel Approach
In stark contrast, the novel methodology disclosed in the patent utilizes a sophisticated palladium-catalyzed cascade reaction that elegantly solves these longstanding issues. By employing a formic acid and acetic anhydride mixture as an in situ carbon monoxide source, the process completely eliminates the need for external CO gas cylinders, drastically simplifying the reactor setup and enhancing operational safety. This one-pot multi-component reaction seamlessly integrates trifluoroethylimidoyl chloride, propargylamine, and acrylamide derivatives to forge the complex carbonyl-bridged biheterocyclic architecture in a single vessel. The reaction proceeds with high efficiency and excellent substrate compatibility, tolerating a wide range of electronic and steric environments on the aromatic rings. This approach not only streamlines the synthetic route by reducing the number of unit operations but also significantly improves the overall atom economy. The ability to generate diverse substituted double heterocyclic compounds through simple substrate design provides medicinal chemists with a powerful tool for rapid structure-activity relationship (SAR) studies without the burden of complex synthesis.
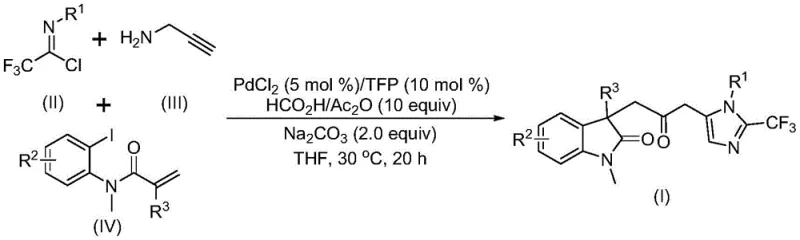
The general reaction scheme illustrates the convergence of three distinct components into a single, highly functionalized product. The use of trifluoroethylimidoyl chloride introduces the valuable trifluoromethyl group, a pharmacophore known to enhance metabolic stability and lipophilicity in drug candidates. The mild reaction temperature of 30°C ensures that thermally labile groups remain intact, while the use of common organic solvents like tetrahydrofuran (THF) facilitates easy workup and solvent recovery. This novel approach transforms a potentially hazardous and tedious multi-step synthesis into a streamlined, safe, and commercially viable process, offering substantial advantages for the manufacturing of complex pharmaceutical intermediates.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
Understanding the mechanistic underpinnings of this transformation is crucial for R&D teams aiming to optimize the process for specific targets. The reaction is believed to initiate with the oxidative addition of a zero-valent palladium species into the carbon-iodine bond of the acrylamide derivative. This is followed by an intramolecular Heck-type reaction, which generates a divalent alkyl-palladium intermediate and establishes the initial cyclic framework. Subsequently, the carbon monoxide released from the decomposition of the formic acid and acetic anhydride mixture inserts into the palladium-carbon bond, forming a key acyl-palladium intermediate. This carbonylation step is the cornerstone of the bridge formation, effectively linking the heterocyclic fragments. Concurrently, the base-promoted reaction between trifluoroethylimidoyl chloride and propargylamine generates a trifluoroacetamidine species, which undergoes isomerization. The final cyclization is driven by the activation of this amidine compound by the acyl-palladium intermediate, leading to the formation of the imidazole ring and the release of the final carbonyl-bridged biheterocyclic product. This intricate dance of organometallic steps occurs in a concerted manner, ensuring high selectivity and minimizing the formation of side products.
From an impurity control perspective, the mechanism offers inherent advantages. The use of a specific ligand system, such as trifurylphosphine (TFP), stabilizes the palladium center and directs the regioselectivity of the insertion steps, preventing the formation of regioisomers that are common in uncatalyzed thermal reactions. The mild conditions prevent the degradation of the trifluoromethyl group and the amide linkage, which are susceptible to hydrolysis or elimination under harsher acidic or basic conditions found in older methods. Furthermore, the stoichiometric balance of reagents, particularly the excess of propargylamine and acrylamide relative to the imidoyl chloride, drives the equilibrium towards the desired product, suppressing the accumulation of unreacted starting materials. The purification process, typically involving filtration and silica gel column chromatography, effectively removes palladium residues and phosphine oxides, ensuring the final product meets the stringent purity specifications required for pharmaceutical applications. This level of mechanistic control translates directly into a cleaner crude reaction profile, reducing the burden on downstream purification units.
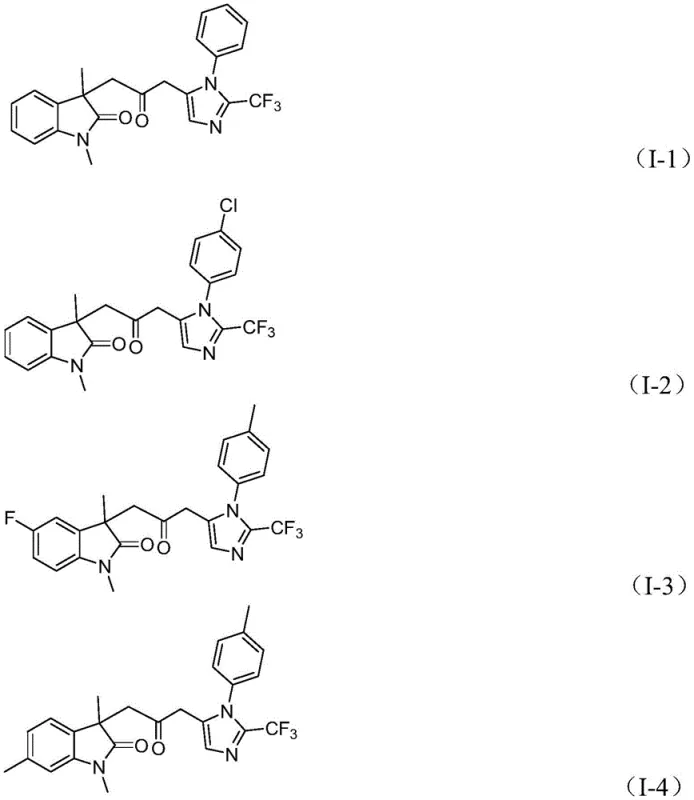
How to Synthesize Carbonyl-Bridged Biheterocyclic Compounds Efficiently
The practical execution of this synthesis is designed for reproducibility and ease of handling in both laboratory and pilot plant settings. The protocol involves charging a reaction vessel with the palladium catalyst, ligand, base, and the CO surrogate mixture in an aprotic solvent. The three organic components are then added, and the mixture is stirred at a controlled low temperature. The simplicity of the operation belies the complexity of the chemical transformations occurring within the flask. Detailed standardized synthesis steps, including precise molar ratios and workup procedures, are outlined below to ensure consistent results across different batches.
- Combine palladium chloride catalyst, trifurylphosphine ligand, sodium carbonate base, and a formic acid/acetic anhydride mixture in an organic solvent like THF.
- Add trifluoroethylimidoyl chloride, propargylamine, and the specific acrylamide derivative to the reaction mixture under stirring.
- Maintain the reaction at 30°C for 12 to 20 hours, then filter and purify the resulting carbonyl-bridged biheterocyclic compound via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this technology offers compelling economic and logistical benefits that extend beyond mere chemical yield. The shift away from toxic carbon monoxide gas to a liquid CO surrogate fundamentally alters the safety profile of the manufacturing site, potentially lowering insurance premiums and reducing the need for specialized gas handling infrastructure. This simplification of the process equipment directly correlates to reduced capital expenditure and faster commissioning times for new production lines. Moreover, the starting materials identified in the patent, such as trifluoroethylimidoyl chloride and propargylamine, are described as cheap and readily available commercial commodities. This availability ensures a stable supply chain, mitigating the risk of raw material shortages that often plague the production of exotic intermediates. The robustness of the reaction conditions, operating at near-ambient temperatures, also implies lower energy costs for heating and cooling compared to processes requiring cryogenic conditions or high-temperature reflux.
- Cost Reduction in Manufacturing: The elimination of toxic gas handling systems and the use of inexpensive, commercially available starting materials significantly lower the overall production costs. By consolidating multiple synthetic steps into a single one-pot reaction, the process reduces labor hours, solvent consumption, and waste disposal fees associated with intermediate isolations. The high reaction efficiency and good yields reported across various substrates mean that less raw material is wasted, further driving down the cost per kilogram of the final active pharmaceutical ingredient precursor. Additionally, the use of a palladium catalyst, while a precious metal, is employed at low loading levels, and the potential for catalyst recovery or the use of cheaper palladium sources like palladium chloride helps maintain cost effectiveness.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals rather than custom-synthesized, unstable intermediates enhances the resilience of the supply chain. Since the key building blocks can be sourced from multiple vendors, the risk of supply disruption is minimized. The scalability of the method, demonstrated by its successful expansion to gram-scale reactions in the patent data, suggests that transitioning to multi-kilogram or ton-scale production is feasible without encountering significant engineering hurdles. This scalability ensures that the supplier can meet fluctuating demand volumes without compromising on quality or lead times, providing a reliable partner for long-term drug development projects.
- Scalability and Environmental Compliance: The process aligns well with green chemistry principles by avoiding hazardous reagents and minimizing waste generation. The use of a liquid CO source instead of gas reduces the risk of atmospheric emissions, aiding in compliance with increasingly strict environmental regulations. The simplified workup procedure, involving filtration and chromatography, is amenable to standard industrial purification techniques, facilitating a smooth scale-up. The ability to tolerate diverse functional groups means that the same platform technology can be applied to synthesize a library of analogs, maximizing the return on investment for the manufacturing facility and reducing the time-to-market for new drug candidates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding this synthesis method, derived from the detailed experimental data and beneficial effects described in the patent documentation. These insights are intended to clarify the operational parameters and strategic value of the technology for potential partners and licensees.
Q: What are the safety advantages of this carbonylation method compared to traditional CO gas methods?
A: This method utilizes a formic acid and acetic anhydride mixture as a safe carbon monoxide substitute, eliminating the need for handling toxic and high-pressure CO gas cylinders, thereby significantly reducing facility safety requirements and operational risks.
Q: What is the substrate compatibility for the R1, R2, and R3 groups in this synthesis?
A: The process demonstrates excellent functional group tolerance, accommodating alkyl, substituted aryl (with methyl, methoxy, halogen, trifluoromethyl, or nitro groups), and benzyl substituents, allowing for the diverse synthesis of drug-like scaffolds.
Q: Is this palladium-catalyzed protocol suitable for large-scale industrial production?
A: Yes, the patent explicitly confirms that the method has been expanded to gram-scale reactions with high efficiency and simple post-treatment, indicating strong potential for commercial scale-up in pharmaceutical intermediate manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Carbonyl-Bridged Biheterocyclic Compounds Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this palladium-catalyzed carbonylation technology in accelerating drug discovery and development. As a leading CDMO expert, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from the bench to the plant. Our state-of-the-art facilities are equipped to handle sensitive organometallic reactions safely, and our rigorous QC labs enforce stringent purity specifications to guarantee the quality of every batch. We understand that consistency and reliability are paramount in the pharmaceutical supply chain, and our dedicated team is committed to delivering high-purity intermediates that meet global regulatory standards.
We invite you to collaborate with us to leverage this advanced synthesis route for your specific pipeline needs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating how this efficient method can optimize your budget. Please contact us to request specific COA data for related compounds or to discuss route feasibility assessments for your target molecules. Together, we can navigate the complexities of modern chemical synthesis and bring life-saving therapies to patients faster and more economically.