Scalable Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles for Advanced Pharmaceutical Intermediates
Scalable Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles for Advanced Pharmaceutical Intermediates
The pharmaceutical and agrochemical industries are constantly seeking robust, scalable, and cost-effective routes to access privileged heterocyclic scaffolds. Among these, the 1,2,4-triazole ring system stands out as a critical pharmacophore found in numerous blockbuster drugs, including antifungal agents like fluconazole and aromatase inhibitors like letrozole. The introduction of a trifluoromethyl group into this heterocyclic framework further enhances the biological profile of the resulting molecules by improving metabolic stability, lipophilicity, and bioavailability. Patent CN110467579B discloses a groundbreaking preparation method for 5-trifluoromethyl substituted 1,2,4-triazole compounds that addresses many of the historical challenges associated with their synthesis. This technology represents a significant leap forward for any organization acting as a reliable pharmaceutical intermediate supplier, offering a pathway to high-purity heterocycles without the burden of complex catalytic systems or harsh reaction conditions.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of nitrogen-containing heterocycles bearing trifluoromethyl groups has been fraught with synthetic difficulties that hinder efficient commercial scale-up of complex pharmaceutical intermediates. Traditional literature reports generally categorize existing methods into two main streams, both of which present substantial drawbacks for large-scale manufacturing. The first approach involves the direct trifluoromethylation of pre-synthesized nitrogen-containing heterocycles. This route typically necessitates the use of specialized, often expensive, and sometimes hazardous trifluoromethylating reagents, which can drive up the cost of goods significantly and introduce safety risks during handling. The second mainstream method relies on reacting synthons bearing a trifluoromethyl group with suitable coupling substrates. While reagents like trifluorodiazoethane are known, they are often unstable or difficult to handle safely on a multi-kilogram scale. Furthermore, the alternative synthon, trifluoroethylimide acid halide, has seen relatively limited application in direct heterocycle formation due to issues with reactivity control and the frequent requirement for transition metal catalysts that leave behind toxic residues requiring extensive purification.
The Novel Approach
In stark contrast to these legacy techniques, the methodology described in patent CN110467579B offers a streamlined, metal-free alternative that drastically simplifies the supply chain for these valuable intermediates. The core innovation lies in the use of inexpensive and readily available hydrazones and trifluoroethylimidoyl chloride as the primary building blocks. This reaction is promoted by elemental iodine, a non-metallic promoter that is both cost-effective and easy to remove during workup compared to heavy metals. The process operates under remarkably mild conditions, specifically heating to 80°C in a common organic solvent like dichloroethane (DCE), and crucially, it does not require stringent anhydrous or anaerobic conditions. This relaxation of environmental controls translates directly to reduced operational complexity and lower infrastructure costs for manufacturing facilities. By eliminating the need for exotic catalysts and sensitive reagents, this novel approach provides a robust platform for cost reduction in API manufacturing, ensuring a more stable and predictable supply of critical triazole intermediates.
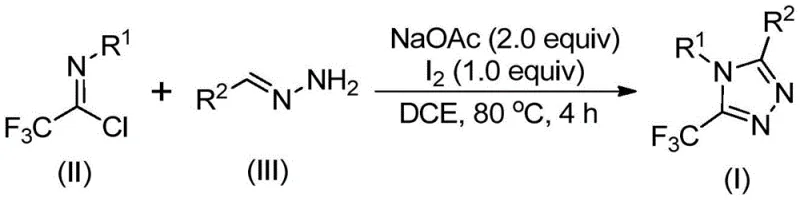
Mechanistic Insights into Iodine-Promoted Cyclization
For R&D directors focused on impurity profiles and process robustness, understanding the mechanistic underpinnings of this transformation is vital. The reaction is believed to proceed through a sophisticated yet efficient cascade initiated by base promotion. Initially, sodium acetate facilitates an intermolecular carbon-nitrogen bond formation between the trifluoroethylimidoyl chloride and the hydrazone, generating a trifluoroacetamidine intermediate. This species then undergoes isomerization to align the reactive centers for cyclization. The subsequent addition of elemental iodine serves a dual purpose: it acts as an oxidant and an iodinating agent. The base-promoted oxidative iodination generates a key iodinated intermediate, which is primed for the final ring-closing event. This is followed by an intramolecular electrophilic substitution reaction that closes the five-membered triazole ring. Finally, aromatization occurs to yield the thermodynamically stable 5-trifluoromethyl substituted 1,2,4-triazole product. This mechanism avoids the radical pathways often associated with trifluoromethylation, leading to cleaner reaction profiles and fewer side products.
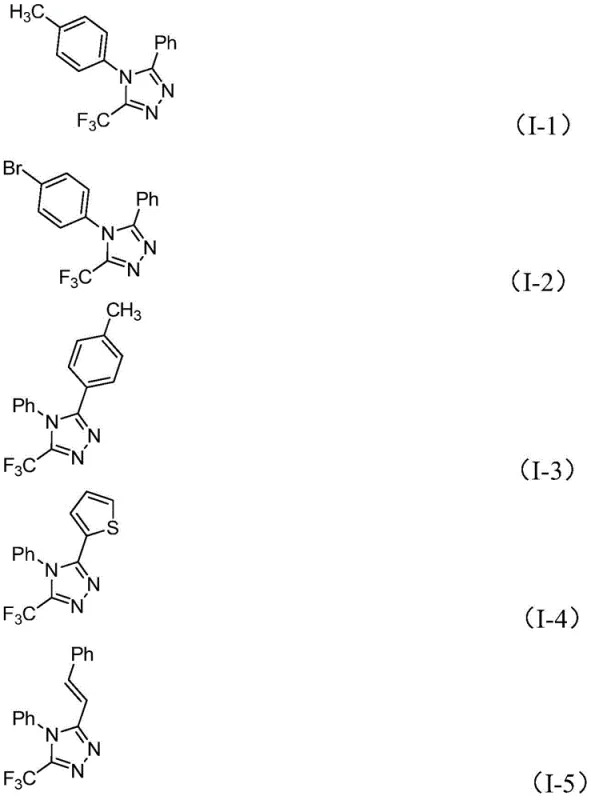
The beauty of this mechanism lies in its broad substrate tolerance, which is essential for generating diverse libraries of analogs during drug discovery. The electronic nature of the substituents on both the imidoyl chloride (R1) and the hydrazone (R2) is well-accommodated. Whether the aryl rings bear electron-donating groups like methyl or methoxy, or electron-withdrawing groups like bromo or nitro, the reaction proceeds with high efficiency. Even heteroaryl groups, such as thiophene or pyridine derivatives, are compatible, as demonstrated by the successful synthesis of various substituted examples. This versatility ensures that the process can be adapted to synthesize a wide array of high-purity OLED material precursors or pharmaceutical intermediates without needing to re-optimize the core reaction conditions for every new substrate, thereby accelerating the timeline from benchtop to pilot plant.
How to Synthesize 5-Trifluoromethyl-1,2,4-Triazole Efficiently
Implementing this synthesis in a laboratory or pilot plant setting is straightforward, leveraging standard equipment and reagents that are globally sourced. The protocol begins with the charging of sodium acetate, trifluoroethylimidoyl chloride, and the specific hydrazone derivative into a reaction vessel containing an aprotic organic solvent, with dichloroethane being the preferred medium for optimal conversion rates. The mixture is heated to approximately 80°C and maintained for an initial period to allow the condensation to occur. Following this, elemental iodine is introduced to the system to drive the oxidative cyclization to completion. The simplicity of the workup, involving filtration and standard silica gel chromatography, further underscores the practicality of this method for producing research quantities or larger batches. For detailed standardized operating procedures and specific stoichiometric ratios tailored to your specific substrate, please refer to the comprehensive guide below.
- Mix sodium acetate, trifluoroethylimidoyl chloride, and hydrazone in an organic solvent such as dichloroethane (DCE).
- Heat the reaction mixture to 80°C and stir for 2 to 4 hours to allow initial condensation and cyclization.
- Add elemental iodine to the system and continue heating for an additional 1 to 2 hours to promote oxidative aromatization.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of this iodine-promoted synthesis route offers compelling strategic advantages that go beyond mere technical feasibility. The primary driver for value creation here is the drastic simplification of the raw material basket. By relying on commodity chemicals like hydrazones (derived from abundant aldehydes) and trifluoroethylimidoyl chloride, manufacturers can mitigate the risks associated with sourcing specialized, single-source fluorinating agents. This shift significantly enhances supply chain reliability, as the upstream availability of these precursors is far more stable than that of exotic trifluoromethyl sources. Furthermore, the elimination of heavy metal catalysts removes the need for expensive and time-consuming metal scavenging steps during purification. This not only reduces the consumption of auxiliary materials but also shortens the overall production cycle time, allowing for faster turnover and improved responsiveness to market demand fluctuations.
- Cost Reduction in Manufacturing: The economic implications of this process are profound, primarily driven by the replacement of costly reagents with commodity chemicals. The use of elemental iodine as a promoter instead of precious metal catalysts like palladium or copper eliminates a major cost center and removes the regulatory burden associated with heavy metal limits in final drug substances. Additionally, the ability to run the reaction without strict anhydrous or anaerobic conditions reduces the energy load and capital expenditure required for specialized reactor setups. These factors combine to deliver substantial cost savings in the production of these high-value intermediates, making the final API more competitive in the global marketplace.
- Enhanced Supply Chain Reliability: Supply continuity is a critical metric for any procurement manager, and this technology bolsters resilience by diversifying the source of the trifluoromethyl group. Since the key building blocks are synthesized from widely available aromatic amines and aldehydes, the risk of supply disruption due to the shortage of a niche reagent is minimized. The robustness of the reaction conditions also means that production is less susceptible to variations in environmental controls, ensuring consistent output quality even in facilities with varying levels of infrastructure sophistication. This reliability is essential for maintaining long-term contracts and meeting the rigorous delivery schedules demanded by top-tier pharmaceutical clients.
- Scalability and Environmental Compliance: As the industry moves towards greener chemistry, the environmental profile of a synthesis route is increasingly important. This method scores highly on sustainability metrics due to its atom economy and the avoidance of toxic heavy metals. The post-treatment process is simple, involving filtration and chromatography, which generates less hazardous waste compared to processes requiring complex aqueous workups to remove metal salts. The patent explicitly notes the ease of scaling this method from gram to industrial levels, suggesting that the heat transfer and mixing requirements are manageable in large reactors. This scalability ensures that the technology can grow with the product lifecycle, from early clinical trials to commercial launch, without the need for disruptive process changes.
Frequently Asked Questions (FAQ)
To assist our partners in evaluating this technology for their specific pipelines, we have compiled answers to common technical inquiries based on the patent data. These questions address the practical aspects of implementation, substrate scope, and purification, providing a clear picture of what to expect when adopting this synthesis route. Understanding these details is crucial for project planning and risk assessment.
Q: What are the key advantages of this iodine-promoted method over traditional trifluoromethylation?
A: This method avoids the use of expensive and hazardous trifluoromethylating reagents or heavy metal catalysts. It utilizes cheap, readily available starting materials like hydrazones and trifluoroethylimidoyl chloride under mild conditions without requiring strict anhydrous or anaerobic environments.
Q: Can this synthesis method be scaled for industrial production?
A: Yes, the patent explicitly states that the method is easy to operate and can be easily expanded to the gram level and beyond, providing significant potential for industrial large-scale production applications due to its simple post-treatment processes.
Q: What types of substituents are tolerated in this triazole synthesis?
A: The method exhibits a wide functional group tolerance. R1 can be various substituted aryl groups (methyl, methoxy, bromo, trifluoromethyl), and R2 can be alkenyl, aryl, or heteroaryl groups, allowing for the design of diverse 1,2,4-triazole derivatives.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-1,2,4-Triazole Supplier
The technological potential of patent CN110467579B is immense, offering a clear path to high-quality trifluoromethylated triazoles that are essential for modern drug discovery. At NINGBO INNO PHARMCHEM, we pride ourselves on our ability to translate such innovative academic and patent literature into robust, commercial-grade manufacturing processes. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can seamlessly transition from the lab bench to the marketplace. Our facility is equipped with rigorous QC labs and adheres to stringent purity specifications, guaranteeing that every batch of intermediate meets the exacting standards required for pharmaceutical applications.
We invite you to explore how this efficient synthesis route can optimize your supply chain and reduce your overall cost of goods. Our technical team is ready to conduct a Customized Cost-Saving Analysis tailored to your specific molecule, evaluating the feasibility of adapting this iodine-promoted method to your unique substrate requirements. Please contact our technical procurement team today to request specific COA data for similar compounds and to discuss route feasibility assessments. Let us collaborate to bring your next generation of therapeutics to patients faster and more economically.