Advanced C-H Activation Strategy for High-Purity Amino Acid Derivatives and Commercial Scale-Up
Advanced C-H Activation Strategy for High-Purity Amino Acid Derivatives and Commercial Scale-Up
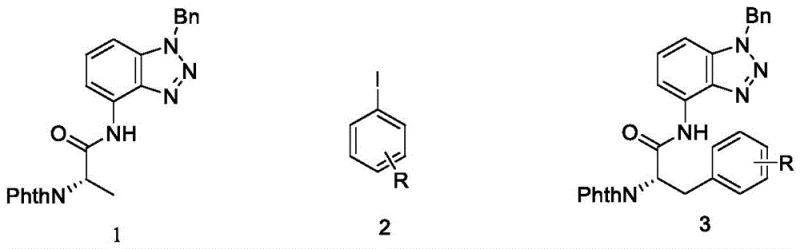
The pharmaceutical industry continuously seeks efficient pathways to construct non-natural alpha-amino acids, which serve as critical building blocks for bioactive peptides and protein engineering. Patent CN116425726A introduces a groundbreaking methodology for synthesizing amino acid derivatives through hydrocarbon activation, specifically targeting the modification of side-chain C(sp3)-H bonds. This technology leverages a novel 4-aminobenzotriazole directing group to achieve precise regioselectivity and stereoselectivity, overcoming the limitations of traditional asymmetric synthesis. By utilizing a palladium-catalyzed system with silver additives, this process enables the direct functionalization of protected amino acid precursors with aryl iodides. For R&D directors and procurement specialists, this represents a significant shift towards more atom-economical and operationally simple manufacturing protocols for high-value intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of chiral alpha-amino acid derivatives has relied heavily on asymmetric hydrogenation or the resolution of racemic mixtures, both of which present substantial economic and technical bottlenecks. Asymmetric synthesis often requires expensive chiral ligands and stringent anhydrous conditions, driving up the cost of goods significantly while limiting substrate scope. Alternatively, chiral resolution methods inherently suffer from a maximum theoretical yield of 50%, generating massive amounts of waste material that must be disposed of or recycled, thereby complicating the supply chain and environmental compliance. Furthermore, direct modification of existing natural amino acids is frequently hampered by the lack of suitable directing groups that can withstand harsh reaction conditions while maintaining stereochemical integrity. These traditional approaches often fail to provide the structural diversity required for modern drug discovery programs, particularly when attempting to introduce bulky aryl groups at the beta-position of the amino acid backbone.
The Novel Approach
The methodology disclosed in CN116425726A circumvents these issues by employing a transition metal-catalyzed C-H activation strategy that directly functionalizes the beta-carbon of phthalimide-protected alanine derivatives. The core innovation lies in the use of 4-aminobenzotriazole as a bidentate directing group, which exhibits stronger coordination capacity and thermal stability compared to conventional guides like 8-aminoquinoline. This robust coordination allows the palladium catalyst to selectively activate the C(sp3)-H bond under relatively mild conditions (80°C), avoiding the decomposition of sensitive functional groups. The reaction utilizes readily available aryl iodides as coupling partners, expanding the chemical space accessible to medicinal chemists without the need for pre-functionalized organometallic reagents. This approach not only simplifies the synthetic route but also enhances the overall efficiency of producing complex amino acid scaffolds essential for next-generation therapeutics.
Mechanistic Insights into Pd-Catalyzed C-H Activation
The catalytic cycle initiates with the coordination of the palladium(II) species to the nitrogen atoms of the 4-aminobenzotriazole moiety on Compound 1, forming a stable five-membered palladacycle intermediate. This cyclometallation step is crucial as it brings the metal center into close proximity with the target beta-C-H bond, lowering the activation energy required for cleavage. The presence of the silver carbonate additive plays a dual role: it acts as a base to facilitate the deprotonation of the C-H bond and serves as an oxidant to regenerate the active palladium species. Following C-H activation, the aryl iodide (Compound 2) undergoes oxidative addition to the palladium center, likely forming a high-valent Pd(IV) intermediate. Subsequent reductive elimination releases the final amino acid derivative (Compound 3) and regenerates the Pd(II) catalyst, completing the cycle. The use of hexafluoroisopropanol (HFIP) as the solvent is particularly strategic, as its high ionizing power stabilizes cationic intermediates and enhances the electrophilicity of the palladium center.
Impurity control in this process is inherently managed by the high specificity of the directing group. Unlike radical-based C-H functionalization which can lead to indiscriminate oxidation at multiple sites, the coordination-directed mechanism ensures that activation occurs exclusively at the beta-position relative to the amide nitrogen. The phthalimide protecting group further shields the alpha-amino center from racemization, preserving the optical purity of the starting material throughout the transformation. Potential side reactions, such as homocoupling of the aryl iodide, are minimized by the precise stoichiometry of the silver additive and the controlled temperature profile. The resulting crude product typically requires only standard silica gel chromatography for purification, indicating a clean reaction profile with minimal byproduct formation. This level of selectivity is vital for meeting the rigorous purity specifications demanded by regulatory bodies for pharmaceutical intermediates.
How to Synthesize Amino Acid Derivatives Efficiently
The synthesis protocol outlined in the patent provides a robust framework for laboratory and pilot-scale production, emphasizing reproducibility and safety. The process begins with the conversion of the protected amino acid to an acid chloride, followed by amidation to install the directing group, and concludes with the key C-H activation step. Each stage is optimized to maximize yield while minimizing operational complexity, making it suitable for adoption by contract development and manufacturing organizations. The detailed standardized synthesis steps below outline the precise reagent ratios, temperatures, and workup procedures required to achieve consistent results.
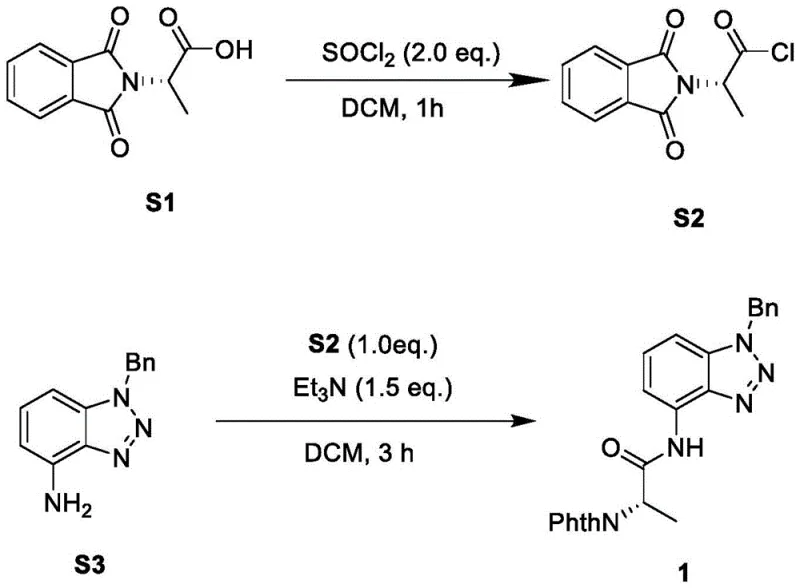
- Convert Phth-protected amino acid (S1) to acid chloride (S2) using thionyl chloride in DCM.
- React acid chloride S2 with 4-aminobenzotriazole and triethylamine to form the directing group precursor (Compound 1).
- Perform Pd-catalyzed C-H activation with aryl iodide (Compound 2) using Ag2CO3 additive in HFIP at 80°C for 24 hours.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this technology offers compelling advantages for procurement managers seeking to optimize the cost structure of amino acid derivative manufacturing. The elimination of expensive chiral ligands and the avoidance of resolution steps translate directly into reduced raw material costs and simplified inventory management. By utilizing commodity chemicals such as palladium acetate and silver carbonate, the process relies on a supply chain that is resilient and widely established, reducing the risk of sourcing bottlenecks. The mild reaction conditions (80°C) allow for the use of standard glass-lined or stainless steel reactors without the need for specialized high-pressure or cryogenic equipment, significantly lowering capital expenditure requirements for scale-up. Furthermore, the high atom economy of the C-H activation approach minimizes waste generation, aligning with increasingly stringent environmental regulations and reducing disposal costs.
- Cost Reduction in Manufacturing: The primary driver for cost optimization in this process is the direct utilization of aryl iodides, which are generally more affordable and stable than corresponding organoboron or organozinc reagents used in cross-coupling. By bypassing the need for pre-functionalization of the amino acid side chain, the number of synthetic steps is drastically reduced, leading to cumulative savings in labor, solvents, and energy consumption. The high efficiency of the 4-aminobenzotriazole directing group ensures high conversion rates, maximizing the throughput of existing reactor volumes. Additionally, the ability to recover and recycle the phthalimide protecting group post-synthesis could further enhance the economic viability of the process on a multi-ton scale.
- Enhanced Supply Chain Reliability: The reliance on commercially available starting materials, such as protected alanine and substituted iodobenzenes, ensures a stable supply chain不受 geopolitical disruptions affecting exotic reagents. The reaction tolerates a wide range of substituents on the aryl ring, providing flexibility to switch between different suppliers of aryl iodides based on price and availability without compromising the reaction outcome. The use of nitrogen as the inert gas atmosphere is standard practice in the fine chemical industry, requiring no specialized infrastructure beyond basic sparging lines. This operational simplicity reduces the dependency on highly skilled operators, mitigating the risk of human error and ensuring consistent batch-to-batch quality.
- Scalability and Environmental Compliance: The process demonstrates excellent scalability potential due to the absence of hazardous reagents like diazomethane or strong alkyl lithium bases. The solvent system, primarily HFIP, can be recovered and distilled for reuse, minimizing volatile organic compound (VOC) emissions. The solid byproducts, mainly silver salts, can be processed for metal recovery, contributing to a circular economy model within the manufacturing facility. The mild thermal profile reduces the energy load on heating and cooling systems, supporting sustainability goals. Overall, the streamlined workflow facilitates the commercial scale-up of complex intermediates from kilogram to multi-ton quantities with minimal process redesign.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation of this C-H activation technology in industrial settings. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on reaction parameters and scope. Understanding these nuances is essential for process engineers evaluating the feasibility of integrating this method into existing production lines.
Q: Why is 4-aminobenzotriazole used as the directing group?
A: According to patent CN116425726A, 4-aminobenzotriazole offers superior coordination ability and thermal stability compared to traditional groups like 8-aminoquinoline, allowing for milder reaction conditions and easier removal.
Q: What are the optimal reaction conditions for this C-H activation?
A: The preferred conditions involve using Pd(OAc)2 (10 mol%) as the catalyst, Ag2CO3 (2.0 eq) as the additive, and hexafluoroisopropanol (HFIP) as the solvent at 80°C under a nitrogen atmosphere for 24 hours.
Q: Can this method be scaled for industrial production?
A: Yes, the method uses commercially available reagents and standard inert gas techniques. The mild temperature (80°C) and robust catalyst system support commercial scale-up of complex intermediates without requiring extreme pressure or cryogenic conditions.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Amino Acid Derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of C-H activation technologies in accelerating drug discovery and development. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative academic discoveries are successfully translated into robust manufacturing processes. Our state-of-the-art facilities are equipped to handle palladium-catalyzed reactions with stringent purity specifications, supported by rigorous QC labs that employ advanced analytical techniques to verify product identity and optical purity. We are committed to delivering high-purity amino acid derivatives that meet the exacting standards of the global pharmaceutical industry.
We invite potential partners to engage with our technical procurement team to discuss how this patented methodology can be adapted to your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this direct functionalization route. We encourage you to contact us for specific COA data and route feasibility assessments, allowing us to demonstrate our capability to support your supply chain with reliable, cost-effective, and high-quality chemical solutions.