Scalable FeCl3-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial API Manufacturing
Scalable FeCl3-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial API Manufacturing
The pharmaceutical and agrochemical industries continuously seek robust synthetic methodologies to access nitrogen-containing heterocycles, particularly those incorporating fluorine motifs which enhance metabolic stability and bioavailability. A pivotal advancement in this domain is detailed in Chinese Patent CN111675662A, which discloses a highly efficient preparation method for 2-trifluoromethyl substituted quinazolinone compounds. This technology addresses critical bottlenecks in heterocyclic chemistry by utilizing a tandem cyclization strategy catalyzed by inexpensive iron salts. For R&D directors and procurement specialists, this represents a shift away from precious metal dependency toward sustainable, cost-effective base metal catalysis. The quinazolinone scaffold is ubiquitous in medicinal chemistry, serving as the core structure for numerous bioactive agents ranging from anticancer to antifungal drugs. By streamlining the introduction of the trifluoromethyl group—a key pharmacophore—this invention provides a reliable pathway for generating high-value chemical libraries and commercial intermediates.
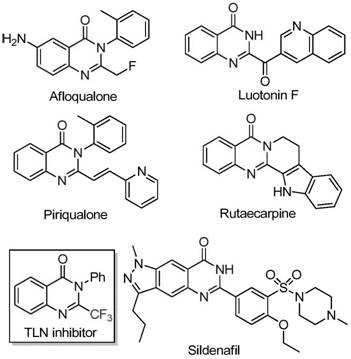
The limitations of conventional methods vs. the novel approach highlight a significant technological leap forward in organic synthesis. Historically, the construction of trifluoromethylated quinazolinones has relied heavily on the cyclization of substrates like anthranilamides or isatoic anhydrides with specific trifluoromethyl synthons such as trifluoroacetic anhydride. These legacy processes are frequently plagued by severe reaction conditions, requiring extreme temperatures or strong acids that limit functional group tolerance. Moreover, the starting materials for these traditional routes are often expensive and difficult to source in bulk quantities, leading to narrow substrate scopes and inconsistent yields. Such inefficiencies create substantial barriers for supply chain heads looking to secure consistent volumes of high-purity intermediates for drug development pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Conventional synthetic routes for accessing 2-trifluoromethyl quinazolinones often suffer from poor atom economy and operational complexity. The reliance on activated trifluoroacetylating agents necessitates rigorous exclusion of moisture and often generates stoichiometric amounts of acidic waste, complicating environmental compliance and waste disposal protocols. Furthermore, the harsh conditions required to drive these cyclizations can lead to the decomposition of sensitive functional groups, thereby restricting the diversity of analogues that can be synthesized. For a procurement manager, these factors translate into higher raw material costs, increased safety hazards during manufacturing, and potential delays due to the need for specialized equipment capable of handling corrosive reagents. The low yields typically associated with these older methods further exacerbate the cost per kilogram, making them less attractive for large-scale commercial applications where margin compression is a constant concern.
The Novel Approach
In stark contrast, the methodology described in the patent utilizes a tandem reaction between readily available trifluoroacetimidoyl chlorides and isatin derivatives, catalyzed by ferric chloride. This innovative route operates under relatively mild conditions, employing a two-stage heating protocol that maximizes conversion while minimizing side reactions. The use of sodium hydride as a base promoter facilitates the initial carbon-nitrogen bond formation, setting the stage for the subsequent iron-catalyzed decarbonylation and cyclization. This approach not only broadens the substrate scope to include various substituted aryl groups but also significantly simplifies the operational workflow. By leveraging earth-abundant iron instead of precious metals, the process inherently reduces the cost of goods sold (COGS) and eliminates the regulatory burden associated with heavy metal residue limits in final active pharmaceutical ingredients.
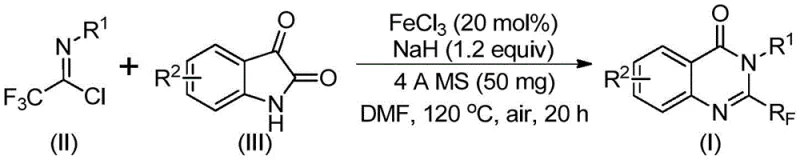
Mechanistic Insights into FeCl3-Catalyzed Tandem Cyclization
The mechanistic pathway proposed for this transformation involves a sophisticated sequence of base-promoted nucleophilic attack followed by transition metal-mediated ring closure. Initially, the sodium hydride deprotonates the isatin nitrogen or activates the imidoyl chloride, fostering an intermolecular carbon-nitrogen bond formation to generate a trifluoroacetamidine intermediate. This step is crucial as it establishes the connectivity required for the heterocyclic core. Subsequently, the ferric chloride catalyst coordinates with the intermediate, facilitating a decarbonylation event that releases carbon monoxide and drives the thermodynamic equilibrium toward the cyclized product. This decarbonylation is a key differentiator from traditional methods, as it allows for the direct utilization of isatin, a cheap and abundant feedstock, rather than requiring pre-functionalized anthranilic acid derivatives. The final isomerization step ensures the formation of the stable quinazolinone aromatic system, resulting in the observed 2-trifluoromethyl substitution pattern with high regioselectivity.
Understanding the impurity profile is essential for R&D teams aiming to replicate this chemistry at scale. The reaction conditions, specifically the use of 4A molecular sieves, play a vital role in scavenging trace water that could otherwise hydrolyze the sensitive imidoyl chloride starting material or deactivate the Lewis acidic iron catalyst. The patent data indicates excellent functional group tolerance, with substituents such as halogens, methyl groups, and methoxy groups remaining intact throughout the vigorous thermal treatment. This robustness suggests that the catalytic cycle is highly selective for the desired C-N and C-C bond formations without promoting unwanted side reactions like homocoupling or over-fluorination. For quality control laboratories, this implies a cleaner crude reaction mixture, which simplifies the purification process and enhances the overall yield of the target high-purity pharmaceutical intermediate.
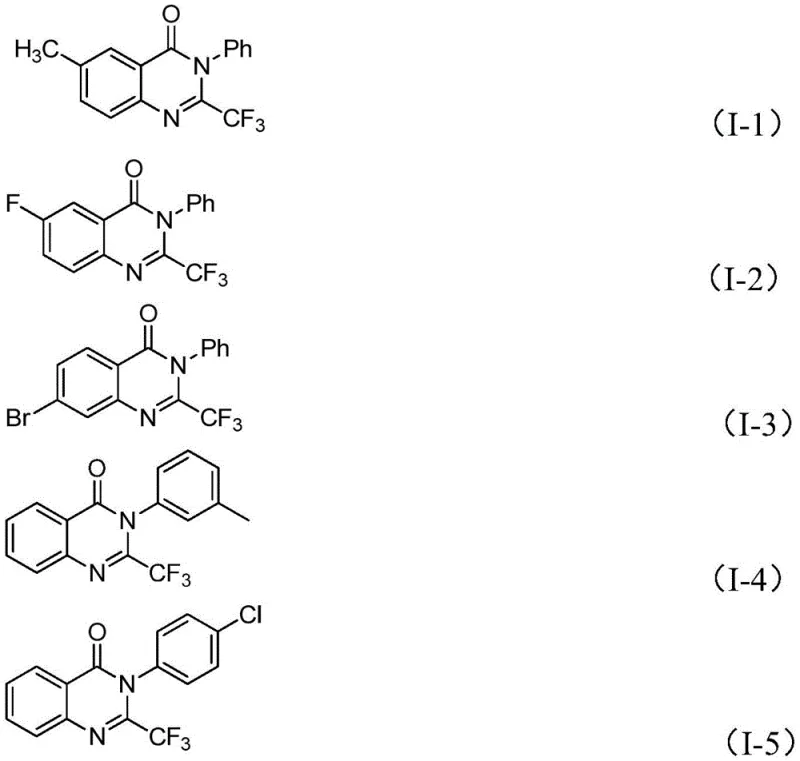
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
The practical execution of this synthesis is designed for reproducibility and ease of handling in standard laboratory or pilot plant settings. The protocol begins with the precise weighing of ferric chloride and sodium hydride, which are combined with 4A molecular sieves in a dry Schlenk tube to ensure an anhydrous environment. The addition of the organic solvent, preferably DMF, creates a homogeneous suspension into which the trifluoroacetimidoyl chloride and isatin derivatives are introduced. The reaction is initiated at a lower temperature of 40°C to allow for the controlled formation of the intermediate species before ramping up to 120°C to drive the cyclization to completion. Detailed standardized synthesis steps for replicating this high-yielding transformation are provided in the guide below.
- Combine ferric chloride (20 mol%), sodium hydride (1.2 equiv), 4A molecular sieves, trifluoroacetimidoyl chloride, and isatin derivative in anhydrous DMF solvent within a Schlenk tube.
- Stir the reaction mixture at 40°C for approximately 10 hours to facilitate the initial intermolecular carbon-nitrogen bond formation.
- Raise the temperature to 120°C and continue stirring for an additional 20 hours under air to complete the decarbonylation and cyclization, followed by filtration and column chromatography purification.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented technology offers compelling advantages that directly address the pain points of modern chemical supply chains. The shift from precious metal catalysts to iron-based systems represents a fundamental change in cost structure, removing the volatility associated with rhodium or palladium pricing. Additionally, the use of commodity chemicals like isatin and simple aromatic amines as precursors ensures a stable and diversified supply base, reducing the risk of single-source dependency. The operational simplicity of the reaction, which proceeds under air after the initial setup, lowers the barrier for adoption by contract manufacturing organizations (CMOs) who may lack specialized inert atmosphere infrastructure. These factors collectively contribute to a more resilient and cost-efficient manufacturing process for complex heterocyclic intermediates.
- Cost Reduction in Manufacturing: The replacement of expensive noble metal catalysts with ferric chloride drastically reduces the direct material costs associated with the catalytic system. Since iron salts are among the cheapest transition metals available, the economic impact on the overall process economics is substantial, especially when scaled to multi-kilogram or tonne production levels. Furthermore, the elimination of costly trifluoroacetic anhydride in favor of easily synthesized imidoyl chlorides further drives down the raw material expenditure. This cost efficiency allows for more competitive pricing strategies when supplying high-purity pharmaceutical intermediates to global markets, enabling better margin management for both manufacturers and end-users.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, including isatin and various substituted anilines, are widely produced commodity chemicals with established global supply chains. This abundance ensures that procurement managers can source raw materials with short lead times and minimal risk of shortage, even during periods of market disruption. The robustness of the reaction conditions also means that the process is less sensitive to minor variations in reagent quality, providing a buffer against supply chain fluctuations. Consequently, manufacturers can maintain consistent production schedules and meet delivery commitments more reliably, which is critical for supporting the continuous clinical trials and commercial launches of new drug candidates.
- Scalability and Environmental Compliance: The protocol has been demonstrated to be scalable to the gram level with high efficiency, indicating a clear path toward kilogram and tonne-scale production without significant re-optimization. The use of DMF as a solvent, while requiring careful handling, is standard in the industry and allows for straightforward recovery and recycling, aligning with green chemistry principles. Moreover, the absence of heavy metal residues simplifies the wastewater treatment process and reduces the environmental footprint of the manufacturing site. This alignment with environmental, social, and governance (ESG) goals is increasingly important for multinational corporations seeking sustainable partners for their API manufacturing needs.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing a factual basis for decision-making. Understanding these nuances helps stakeholders evaluate the feasibility of integrating this method into their existing production workflows or R&D pipelines.
Q: What are the primary advantages of using FeCl3 over precious metal catalysts for quinazolinone synthesis?
A: The use of ferric chloride (FeCl3) offers significant economic advantages as it is an inexpensive, earth-abundant base metal compared to palladium or rhodium catalysts. Furthermore, it eliminates the need for complex ligand systems and reduces the risk of heavy metal contamination in the final pharmaceutical intermediate, simplifying downstream purification and regulatory compliance.
Q: How does this method improve upon traditional trifluoromethylation strategies?
A: Traditional methods often rely on harsh conditions and expensive trifluoroacetic anhydride or ethyl trifluoroacetate synthons with limited substrate scope. This novel approach utilizes readily available trifluoroacetimidoyl chlorides and isatins, operating under milder, two-stage thermal conditions that tolerate a wide range of functional groups including halogens, alkyls, and nitro groups.
Q: Is this synthesis protocol suitable for large-scale industrial production?
A: Yes, the patent explicitly demonstrates scalability to the gram level with high yields ranging up to 93%. The use of common solvents like DMF, commercially available starting materials, and a simple workup procedure involving filtration and silica gel chromatography makes it highly amenable to commercial scale-up for API intermediate manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
As the demand for fluorinated heterocycles continues to grow in the pharmaceutical sector, partnering with an experienced CDMO is essential for navigating the complexities of process development and scale-up. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from the bench to the plant. Our commitment to quality is underscored by our stringent purity specifications and rigorous QC labs, which guarantee that every batch of 2-trifluoromethyl quinazolinone meets the highest international standards for safety and efficacy. We understand the critical nature of timeline adherence in drug development and are equipped to handle the unique challenges of fluorine chemistry with precision and care.
We invite you to engage with our technical team to explore how this innovative iron-catalyzed route can optimize your supply chain and reduce costs. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits specific to your volume requirements. We encourage potential partners to contact our technical procurement team to obtain specific COA data and route feasibility assessments tailored to your project needs. Let us collaborate to bring your next generation of therapeutic agents to market faster and more efficiently.