Advanced Metal-Free Synthesis of Triazolopyridines for Commercial Pharmaceutical Applications
Introduction to Next-Generation Triazolopyridine Synthesis
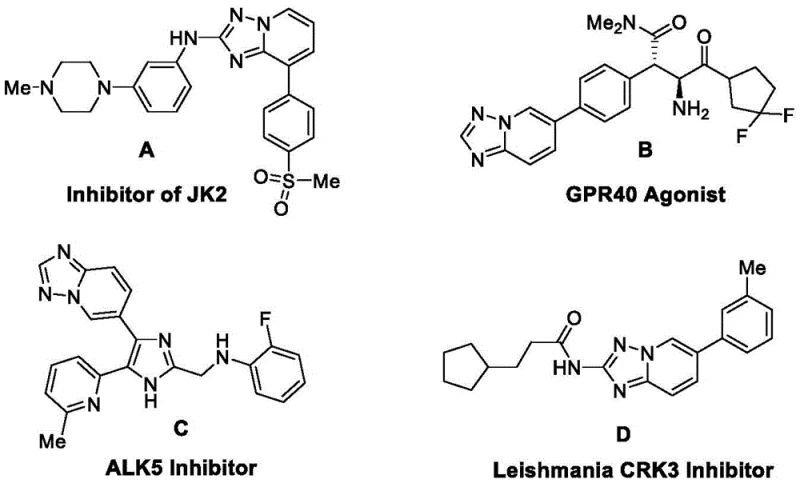
The pharmaceutical industry continuously seeks robust and sustainable pathways for constructing privileged heterocyclic scaffolds, particularly those found in kinase inhibitors and G-protein coupled receptor modulators. Patent CN114644629B, published in June 2023, introduces a groundbreaking synthetic methodology for [1,2,4]triazolo[1,5-a]pyridine compounds that addresses critical inefficiencies in current manufacturing protocols. This core structural unit is prevalent in high-value bioactive molecules, including potent JK2 inhibitors and GPR40 agonists, which are essential for treating metabolic disorders and myeloproliferative neoplasms. The disclosed innovation leverages a direct condensation strategy between 3-amino-4-azaisoxazole derivatives and 2-halogen substituted pyridines, bypassing the need for expensive transition metal catalysts.
By utilizing a base-mediated cyclization in polar aprotic solvents, this technology offers a streamlined route that significantly enhances atom economy and reduces hazardous waste generation. For R&D directors and process chemists, this represents a pivotal shift towards greener chemistry without compromising on yield or purity. The method's ability to tolerate a wide array of functional groups ensures versatility in library synthesis for drug discovery programs. Furthermore, the operational simplicity of the reaction conditions suggests immediate applicability in industrial settings, positioning this patent as a key asset for reliable pharmaceutical intermediate supplier networks aiming to optimize their supply chains.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the [1,2,4]triazolo[1,5-a]pyridine core has relied on oxidative cyclization strategies that pose significant environmental and safety challenges. Pioneering work by Sletzinger and Bhattacharyya in the 1960s utilized N-pyridylamidines with oxidants such as sodium hypochlorite, lead acetate, or manganese dioxide, generating substantial heavy metal waste. Later developments by Nagasawa involved reactions between 2-aminopyridines and organic nitriles, which often required harsh conditions and exhibited limited substrate scope. More recent approaches employing hypervalent iodine reagents, while effective, introduce high material costs and safety concerns regarding the handling of energetic oxidants on a large scale.
These traditional pathways frequently suffer from poor functional group compatibility, necessitating extensive protection and deprotection sequences that inflate production timelines and costs. The reliance on transition metal catalysts also mandates rigorous purification steps to meet stringent residual metal specifications required by regulatory bodies for API manufacturing. Consequently, the industry has faced a persistent bottleneck in scaling these syntheses efficiently, driving up the cost of goods for downstream drug candidates. The accumulation of toxic byproducts and the complexity of workup procedures further exacerbate the environmental footprint, conflicting with modern sustainability goals in fine chemical manufacturing.
The Novel Approach
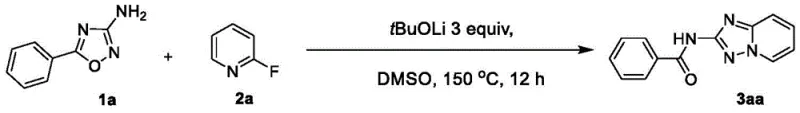
In stark contrast, the methodology described in CN114644629B employs a direct nucleophilic substitution and cyclization cascade that completely eliminates the need for external oxidants or metal catalysts. By reacting 3-amino-4-azaisoxazole derivatives with 2-fluoropyridines or other 2-halo substituted pyridines in the presence of a strong base like lithium tert-butoxide, the reaction proceeds smoothly to form the target heterocycle. This approach not only simplifies the reaction setup but also drastically reduces the impurity profile associated with metal residues and oxidation byproducts. The use of readily available starting materials enhances the economic viability of the process, making it an attractive option for cost reduction in API manufacturing.
The operational parameters are remarkably straightforward, typically involving heating the reaction mixture in dimethyl sulfoxide (DMSO) at temperatures around 150°C for approximately 12 hours. This thermal stability allows for flexible reactor configurations and minimizes the risk of thermal runaway associated with exothermic oxidations. Moreover, the workup procedure is benign, often requiring only simple extraction with ethyl acetate and brine washing followed by column chromatography. This simplicity translates directly into reduced processing time and lower utility consumption, providing a compelling value proposition for supply chain heads focused on efficiency and throughput optimization in high-volume production environments.
Mechanistic Insights into Base-Mediated Cyclization
The underlying mechanism of this transformation involves a sequential nucleophilic aromatic substitution followed by intramolecular cyclization, driven by the strong basicity of reagents such as tBuOLi. Initially, the base deprotonates the amino group of the 3-amino-4-azaisoxazole derivative, generating a highly nucleophilic nitrogen anion. This species then attacks the electron-deficient carbon at the 2-position of the halopyridine ring, displacing the halide leaving group (typically fluoride or chloride) to form an intermediate adduct. The subsequent rearrangement and ring closure are facilitated by the inherent instability of the isoxazole ring under these thermal conditions, leading to the extrusion of oxygen or rearrangement to form the stable triazolopyridine system.
This mechanistic pathway is exceptionally robust against steric hindrance and electronic variations, explaining the broad substrate scope observed in the patent examples. Whether the pyridine ring bears electron-withdrawing groups like chlorine or bromine, or electron-donating groups like methyl or methoxy, the reaction proceeds with consistent efficiency. For process chemists, understanding this mechanism is crucial for troubleshooting and optimizing reaction parameters such as base stoichiometry and solvent choice. The absence of radical intermediates or metal-coordination steps ensures a clean reaction profile, minimizing the formation of difficult-to-remove side products that often plague transition-metal catalyzed cross-coupling reactions.
Furthermore, the tolerance for diverse substituents on the R1 and R2 positions allows for the late-stage functionalization of complex molecules, a key requirement in medicinal chemistry campaigns. The ability to incorporate halogens directly into the scaffold without affecting the cyclization step provides valuable handles for subsequent derivatization via Suzuki or Buchwald-Hartwig couplings. This modularity enhances the strategic value of the intermediates produced, enabling rapid exploration of structure-activity relationships (SAR) while maintaining a streamlined synthetic route. Such mechanistic clarity empowers R&D teams to confidently adapt this chemistry for novel targets beyond the specific examples listed in the patent documentation.
How to Synthesize [1,2,4]triazolo[1,5-a]pyridine Efficiently
To implement this synthesis effectively, precise control over reaction stoichiometry and temperature is essential to maximize yield and minimize impurity formation. The patent outlines a generalized protocol where the amino-isoxazole and halopyridine are combined in a dry polar aprotic solvent, followed by the careful addition of the base under an inert atmosphere. Detailed standardized operating procedures for scaling this reaction from gram to kilogram quantities are critical for ensuring reproducibility and safety. The following guide summarizes the key operational steps derived from the exemplary embodiments provided in the intellectual property disclosure.
- Combine 3-amino-4-azaisoxazole derivative and 2-halogen substituted pyridine in an organic solvent such as DMSO.
- Add a strong base like lithium tert-butoxide (3 equivalents) under a protective gas atmosphere.
- Heat the reaction mixture to 150°C for 12 hours, then cool, extract with ethyl acetate, and purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this synthetic route offers transformative benefits for procurement managers and supply chain leaders tasked with securing reliable sources of high-quality intermediates. The elimination of precious metal catalysts such as palladium or copper removes a significant cost driver and supply risk, as these metals are subject to volatile market pricing and geopolitical supply constraints. Additionally, the avoidance of hazardous oxidants reduces the regulatory burden associated with waste disposal and worker safety, leading to substantial cost savings in environmental compliance and insurance premiums. The simplicity of the raw material supply chain, relying on commodity chemicals rather than specialized reagents, further enhances supply security.
- Cost Reduction in Manufacturing: The removal of transition metal catalysts and expensive oxidants significantly lowers the raw material bill of costs. Furthermore, the simplified workup procedure reduces solvent consumption and processing time, leading to lower utility and labor costs per kilogram of product. The high yields reported in the patent examples, often exceeding 70%, contribute to better overall material efficiency and reduced waste generation. This economic efficiency is vital for maintaining competitive pricing in the generic pharmaceutical market where margin pressure is intense.
- Enhanced Supply Chain Reliability: The starting materials, including various substituted 2-fluoropyridines and amino-isoxazoles, are widely available from multiple global chemical suppliers, reducing dependency on single-source vendors. The robustness of the reaction conditions means that production is less susceptible to minor variations in raw material quality or environmental factors, ensuring consistent output. This reliability is crucial for maintaining continuous manufacturing schedules and meeting tight delivery deadlines for downstream API synthesis partners who depend on just-in-time inventory models.
- Scalability and Environmental Compliance: The use of standard solvents like DMSO and moderate temperatures facilitates easy scale-up from laboratory benchtop to multi-ton industrial reactors without requiring specialized high-pressure or cryogenic equipment. The absence of heavy metal residues simplifies the purification process, ensuring that the final product easily meets stringent international quality standards for pharmaceutical ingredients. This alignment with green chemistry principles also supports corporate sustainability initiatives, enhancing the brand reputation of manufacturers who adopt this eco-friendly technology.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects described in the patent specification, providing clarity for stakeholders evaluating this process for adoption. Understanding these nuances helps in making informed decisions about process integration and resource allocation.
Q: What are the primary advantages of this synthesis method over traditional routes?
A: This method eliminates the need for toxic transition metal catalysts and hazardous oxidants like hypervalent iodine or lead acetate, resulting in a greener process with simplified purification and reduced environmental impact.
Q: Does this protocol support diverse functional groups on the substrate?
A: Yes, the patent demonstrates excellent functional group compatibility, tolerating various substituents such as halogens, alkyl groups, alkoxy groups, and aryl rings on both the isoxazole and pyridine moieties without significant yield loss.
Q: Is this method suitable for large-scale industrial production?
A: The process utilizes commercially available raw materials and standard solvents like DMSO under relatively mild thermal conditions (150°C), making it highly adaptable for commercial scale-up of complex heterocycles without requiring specialized high-pressure equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable [1,2,4]triazolo[1,5-a]pyridine Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient and sustainable synthetic routes in the modern pharmaceutical landscape. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from development to full-scale manufacturing. We are committed to delivering high-purity [1,2,4]triazolo[1,5-a]pyridine derivatives that meet rigorous QC labs standards and stringent purity specifications required by global regulatory agencies. Our state-of-the-art facilities are equipped to handle the specific solvent and thermal requirements of this base-mediated cyclization safely and efficiently.
We invite you to collaborate with us to leverage this innovative technology for your next drug development program. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our capabilities can accelerate your timeline and optimize your budget. Let us be your partner in transforming cutting-edge patent chemistry into commercial reality.