Advanced Palladium-Catalyzed C-H Activation for Scalable Deazapurine Derivative Manufacturing
The pharmaceutical industry continuously seeks efficient pathways to access complex heterocyclic scaffolds, particularly those exhibiting potent biological activity against oncological targets. Patent CN114957262A introduces a groundbreaking methodology for the preparation of C-6 arylated deazapurine derivatives, addressing critical bottlenecks in the synthesis of pyrrolo[2,3-d]pyrimidine cores. This innovation establishes a new synthesis route that directly activates C-H bonds under the catalysis of palladium under remarkably mild reaction conditions, introducing arylation functional groups with exceptional regioselectivity. The technology enables the rapid, simple, and efficient realization of functional group modification on deazapurine derivatives, characterized by straightforward operational procedures, robust substrate universality, and superior reaction selectivity. By expanding the application range of deazapurine compounds, this patent provides a vital tool for developing next-generation kinase inhibitors and anticancer agents, positioning it as a cornerstone technology for reliable pharmaceutical intermediate supplier networks aiming to optimize their API pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the functionalization of pyrrolo[2,3-d]pyrimidine derivatives has been plagued by significant synthetic challenges that hinder efficient commercial scale-up of complex pharmaceutical intermediates. Traditional methods often rely on harsh reaction conditions, including elevated temperatures and the use of aggressive reagents that can degrade sensitive functional groups on the molecular scaffold. Furthermore, many existing protocols suffer from limited substrate scope, failing to accommodate diverse electronic environments required for modern drug discovery libraries. A major drawback in conventional cross-coupling strategies is the necessity for pre-halogenated starting materials, which adds extra synthetic steps, increases raw material costs, and generates substantial halogenated waste streams. Additionally, achieving high regioselectivity at the C-6 position without affecting other reactive sites on the purine mimic has proven difficult, often resulting in complex mixtures of isomers that require tedious and yield-loss-inducing purification processes. These inefficiencies collectively drive up the cost of goods and extend lead times, creating friction in the supply chain for high-purity active pharmaceutical ingredients.
The Novel Approach
In stark contrast to these legacy methods, the patented technology leverages a sophisticated palladium-catalyzed C-H direct activation strategy that fundamentally simplifies the synthetic architecture. This novel approach utilizes a catalytic system comprising palladium acetate or palladium trifluoroacetate in conjunction with TEMPO as a mild oxidant, all dissolved in trifluoroacetic acid. The reaction proceeds efficiently at temperatures ranging from 20°C to 35°C, eliminating the need for energy-intensive heating or cryogenic cooling systems. This method achieves direct arylation at the C-6 position with high precision, bypassing the need for pre-functionalized halides and thereby reducing the overall step count. The robustness of this chemistry is evidenced by its tolerance to a wide array of substituents, including electron-donating and electron-withdrawing groups on the aniline moiety. 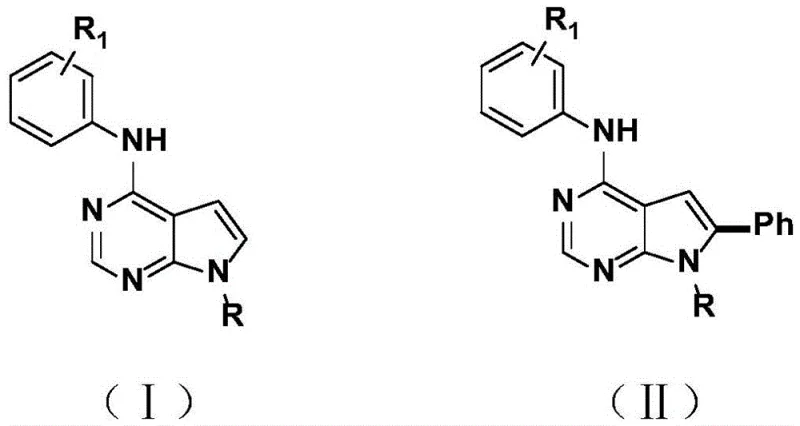 This streamlined process not only enhances the speed of synthesis but also significantly improves the environmental profile of the manufacturing process by reducing waste and energy consumption, aligning perfectly with green chemistry principles essential for cost reduction in API manufacturing.
This streamlined process not only enhances the speed of synthesis but also significantly improves the environmental profile of the manufacturing process by reducing waste and energy consumption, aligning perfectly with green chemistry principles essential for cost reduction in API manufacturing.
Mechanistic Insights into Palladium-Catalyzed C-H Activation
The core of this technological advancement lies in the intricate mechanistic pathway facilitated by the palladium catalyst. The reaction initiates with the coordination of the palladium species to the nitrogen atoms of the pyrrolo[2,3-d]pyrimidine scaffold, which acts as a directing group to guide the metal center to the proximal C-6 position. Under the oxidative conditions provided by TEMPO, the palladium center undergoes a crucial oxidation state change that enables the cleavage of the relatively inert C-H bond. This C-H activation step is the rate-determining factor in many similar transformations, but the specific solvent system of trifluoroacetic acid stabilizes the transition state, lowering the activation energy barrier. Following C-H cleavage, the transmetallation with the phenylboronic acid occurs, transferring the aryl group to the palladium center. Finally, a reductive elimination step releases the desired C-6 arylated product and regenerates the active palladium catalyst, completing the catalytic cycle. This mechanism ensures that the reaction remains highly selective for the C-6 position, avoiding unwanted substitution at the C-2 or N-7 positions which are common side reactions in less optimized systems.
Impurity control is paramount in the production of high-purity pharmaceutical intermediates, and this method demonstrates exceptional capability in minimizing by-product formation. The high regioselectivity inherently reduces the formation of positional isomers, which are often the most difficult impurities to separate chromatographically. Furthermore, the mild oxidative environment prevents the over-oxidation of sensitive functional groups such as sulfides or amines that might be present on the substrate. Experimental data from the patent indicates that the resulting products consistently achieve HPLC purities exceeding 97%, with many examples reaching 98% or higher without extensive recrystallization. 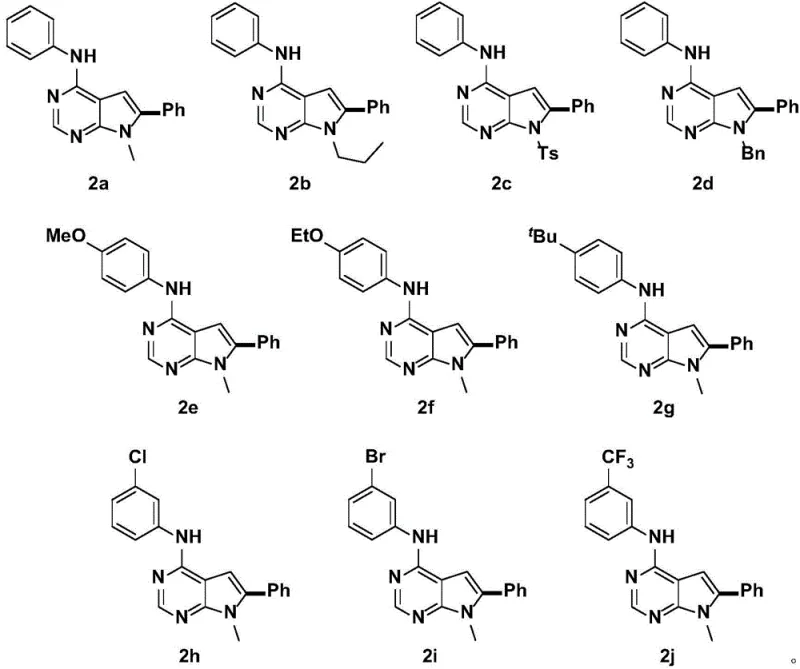 The ability to tolerate diverse substituents, such as methoxy, ethoxy, tert-butyl, chloro, bromo, and trifluoromethyl groups, underscores the versatility of this catalytic system. This broad substrate scope allows medicinal chemists to rapidly generate analog libraries for structure-activity relationship (SAR) studies without being constrained by synthetic feasibility, thereby accelerating the drug discovery timeline.
The ability to tolerate diverse substituents, such as methoxy, ethoxy, tert-butyl, chloro, bromo, and trifluoromethyl groups, underscores the versatility of this catalytic system. This broad substrate scope allows medicinal chemists to rapidly generate analog libraries for structure-activity relationship (SAR) studies without being constrained by synthetic feasibility, thereby accelerating the drug discovery timeline.
How to Synthesize C-6 Arylated Deazapurine Efficiently
Implementing this synthesis route requires careful attention to reagent stoichiometry and reaction monitoring to maximize yield and purity. The standard protocol involves dissolving the starting deazapurine derivative, phenylboronic acid, TEMPO, and the palladium catalyst in trifluoroacetic acid at a specific molar ratio, typically favoring a slight excess of the boronic acid and oxidant to drive the reaction to completion. The reaction mixture is then stirred at room temperature for a defined period, usually between 3 to 5 hours, after which the progress is monitored via TLC or HPLC. Upon completion, the workup procedure is straightforward, involving quenching with water and pH adjustment to facilitate extraction.  The detailed standardized synthesis steps, including precise quantities, safety precautions, and purification parameters, are outlined below to ensure reproducibility and compliance with GMP standards.
The detailed standardized synthesis steps, including precise quantities, safety precautions, and purification parameters, are outlined below to ensure reproducibility and compliance with GMP standards.
- Dissolve the N-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine substrate, phenylboronic acid, TEMPO oxidant, and palladium catalyst in trifluoroacetic acid solvent.
- Stir the reaction mixture at mild temperatures between 20°C and 35°C for a duration of 3 to 5 hours to ensure complete conversion.
- Quench the reaction with water, adjust pH to 9-10 using sodium hydroxide, extract with ethyl acetate, and purify via flash column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis method offers transformative benefits that directly impact the bottom line and operational resilience. The shift from multi-step halogenation/coupling sequences to a direct C-H activation protocol drastically simplifies the manufacturing workflow, reducing the number of unit operations required. This simplification translates into substantial cost savings by lowering labor hours, reducing solvent consumption, and minimizing the footprint of the production facility. Moreover, the use of commercially available and inexpensive reagents like phenylboronic acid and palladium acetate ensures a stable and predictable raw material supply chain, mitigating the risks associated with sourcing exotic or custom-synthesized starting materials. The mild reaction conditions also enhance equipment longevity and reduce maintenance costs, as reactors are not subjected to extreme thermal stress or corrosive environments typical of traditional harsh chemistries.
- Cost Reduction in Manufacturing: The elimination of pre-functionalization steps removes the need for purchasing or synthesizing expensive halogenated intermediates, which are often cost-prohibitive at scale. By streamlining the synthesis into a single catalytic step, the overall material cost is significantly reduced, and the yield losses associated with isolation of intermediate compounds are completely avoided. The high atom economy of the direct arylation further contributes to waste reduction, lowering disposal costs and improving the overall process mass intensity (PMI). These factors combine to create a highly cost-effective manufacturing route that enhances profit margins without compromising on quality.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals such as trifluoroacetic acid and common boronic acids ensures that the supply chain is robust and less susceptible to geopolitical disruptions or vendor shortages. The short reaction time of 3 to 5 hours allows for faster batch turnover, enabling manufacturers to respond more agilely to fluctuations in market demand. This agility is crucial for maintaining continuous supply to downstream API manufacturers, preventing stockouts and ensuring that clinical trial materials or commercial drugs are delivered on schedule. The scalability of the process from gram to kilogram scales without re-optimization further secures the supply chain against capacity bottlenecks.
- Scalability and Environmental Compliance: Operating at near-ambient temperatures significantly reduces the energy load required for heating or cooling, aligning with global sustainability goals and reducing the carbon footprint of the manufacturing process. The simplified workup procedure, which avoids complex distillations or hazardous quenches, minimizes the generation of hazardous waste streams, facilitating easier compliance with increasingly stringent environmental regulations. The process is inherently safer, reducing the risk of thermal runaways or pressure build-ups, which is a critical consideration for large-scale industrial production. This environmental and safety profile makes the technology attractive for contract manufacturing organizations (CMOs) looking to expand their portfolio of green chemistry capabilities.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this C-H activation technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on the practical aspects of adopting this synthesis route. Understanding these details is essential for technical teams evaluating the feasibility of integrating this method into their existing production lines or R&D workflows.
Q: What represents the primary advantage of this C-H activation method over traditional cross-coupling?
A: The primary advantage is the elimination of pre-functionalized halogenated substrates, allowing for direct C-H bond activation at the C-6 position under mild conditions, which significantly reduces synthetic steps and waste generation.
Q: How does the process ensure high regioselectivity for the C-6 position?
A: The process utilizes a specific palladium catalyst system combined with TEMPO oxidation in trifluoroacetic acid, which directs the electrophilic attack specifically to the C-6 position of the pyrrole ring, minimizing isomeric by-products.
Q: Is this synthesis method suitable for large-scale commercial production?
A: Yes, the method operates at near-room temperature (20-35°C) without requiring cryogenic conditions or expensive ligands, making it highly amenable to scale-up for industrial manufacturing of pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Deazapurine Derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic value of advanced synthetic methodologies like the one described in CN114957262A for driving innovation in oncology drug development. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to full-scale manufacturing is seamless and efficient. Our facilities are equipped with state-of-the-art reactor systems capable of handling sensitive palladium-catalyzed reactions under strictly controlled conditions, guaranteeing consistent batch-to-batch quality. We adhere to stringent purity specifications and operate rigorous QC labs to verify that every batch of deazapurine intermediate meets the highest international standards for identity, potency, and impurity profiles.
We invite you to collaborate with us to leverage this cutting-edge technology for your next project. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline constraints. By partnering with us, you gain access to our deep expertise in process optimization and regulatory compliance. Please contact our technical procurement team today to request specific COA data for our catalog compounds or to discuss route feasibility assessments for your custom synthesis needs. Let us help you accelerate your pipeline with reliable, high-quality chemical solutions.