Advanced Rhodium-Catalyzed C-H Activation for Scalable Indole Derivative Manufacturing
Advanced Rhodium-Catalyzed C-H Activation for Scalable Indole Derivative Manufacturing
The pharmaceutical and fine chemical industries are constantly seeking more efficient pathways to construct privileged scaffolds like the indole ring, which is ubiquitous in bioactive molecules ranging from cholesterol-lowering agents like Fluvastatin sodium to anticancer candidates such as Dictyodendrin B. Patent CN111285846B introduces a groundbreaking synthetic methodology that leverages transition metal catalysis to streamline the production of 2-(2-indolyl)-acetate derivatives. This technology utilizes a rhodium-catalyzed C-H activation strategy, bypassing the need for pre-functionalized substrates and offering a direct route to complex heterocycles from readily available N-phenyl-2-aminopyridines and allenoic acid esters. By shifting away from classical stoichiometric methods towards catalytic atom-economical processes, this innovation addresses critical pain points in modern process chemistry, specifically regarding waste generation and step count reduction.
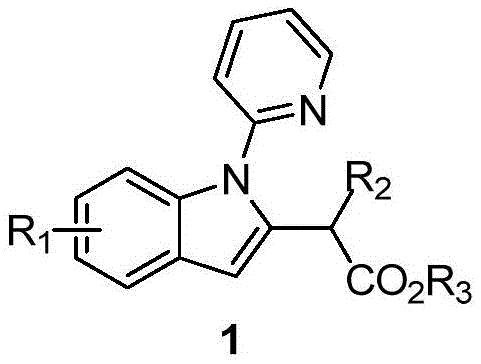
For R&D Directors evaluating new process technologies, the structural versatility offered by this patent is particularly compelling. The core structure allows for significant diversification at three distinct positions: the phenyl ring (R1), the alkyl chain adjacent to the ester (R2), and the ester group itself (R3). This flexibility means that a single robust platform technology can be adapted to generate a wide library of analogs for structure-activity relationship (SAR) studies without necessitating a complete process redevelopment for each new compound. The ability to introduce diverse functional groups such as halogens, cyano, and alkoxy groups directly during the cyclization step significantly accelerates the drug discovery timeline, making this a highly valuable asset for medicinal chemistry campaigns targeting indole-based therapeutics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the construction of the indole nucleus has relied heavily on classical named reactions such as the Fischer, Madelung, or Bischler indole syntheses. While these methods have served the industry for over a century, they suffer from inherent limitations that become problematic in the context of modern green chemistry and large-scale manufacturing. For instance, the Fischer indole synthesis often requires harsh acidic conditions and high temperatures, which can be incompatible with sensitive functional groups present in advanced intermediates. Furthermore, these classical approaches frequently demand pre-functionalized starting materials, such as hydrazines or ortho-substituted anilines, which adds extra synthetic steps, increases raw material costs, and generates substantial amounts of chemical waste. The requirement for stoichiometric reagents and the generation of byproducts like ammonia or water in condensation steps often lead to lower atom economy, forcing process chemists to deal with complex purification challenges and reduced overall yields.
The Novel Approach
In stark contrast, the methodology disclosed in CN111285846B represents a paradigm shift by utilizing direct C-H bond functionalization. This approach activates the ortho-C-H bond of the N-phenyl ring in N-phenyl-2-aminopyridine directly, coupling it with allenoic acid esters to forge the indole core in a single catalytic cycle. 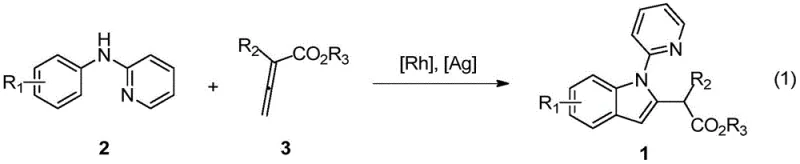 This novel route eliminates the need for pre-halogenation or other activating groups on the aromatic ring, thereby reducing the step count and improving the overall mass balance of the synthesis. The reaction proceeds under relatively mild conditions (optimized at 60°C) compared to the vigorous conditions often required by traditional cyclizations. Moreover, the use of allenoic acid esters as coupling partners introduces a unique reactivity profile, allowing for the installation of functionalized side chains that would be difficult to achieve via standard electrophilic aromatic substitution or cross-coupling protocols, thus expanding the chemical space accessible to process chemists.
This novel route eliminates the need for pre-halogenation or other activating groups on the aromatic ring, thereby reducing the step count and improving the overall mass balance of the synthesis. The reaction proceeds under relatively mild conditions (optimized at 60°C) compared to the vigorous conditions often required by traditional cyclizations. Moreover, the use of allenoic acid esters as coupling partners introduces a unique reactivity profile, allowing for the installation of functionalized side chains that would be difficult to achieve via standard electrophilic aromatic substitution or cross-coupling protocols, thus expanding the chemical space accessible to process chemists.
Mechanistic Insights into Rhodium-Catalyzed C-H Activation
The success of this transformation hinges on the precise orchestration of a Rhodium(III) catalytic cycle, which facilitates the cleavage of the inert C-H bond and subsequent C-C bond formation. The mechanism likely initiates with the coordination of the pyridine nitrogen atom in the N-phenyl-2-aminopyridine substrate to the cationic rhodium species generated in situ from [RhCp*Cl2]2 and AgSbF6. This coordination directs the metal center to the ortho-position of the phenyl ring, enabling the concerted metalation-deprotonation (CMD) or electrophilic metallation pathway to activate the C-H bond. The presence of silver salts like AgOAc serves a dual purpose: acting as an oxidant to regenerate the active Rh(III) species and potentially assisting in the deprotonation step. The resulting rhodacycle intermediate then undergoes migratory insertion with the allenoic acid ester, followed by reductive elimination or protonolysis to release the indole product and close the catalytic cycle.  Understanding this mechanism is crucial for R&D teams as it highlights the importance of the pyridine directing group; removing or modifying this group could disrupt the coordination geometry and halt the reaction, emphasizing the specificity of this catalytic system.
Understanding this mechanism is crucial for R&D teams as it highlights the importance of the pyridine directing group; removing or modifying this group could disrupt the coordination geometry and halt the reaction, emphasizing the specificity of this catalytic system.
From an impurity control perspective, the high regioselectivity of this C-H activation process is a significant advantage. Because the reaction is directed by the intramolecular coordination of the pyridine nitrogen, the activation occurs exclusively at the ortho-position relative to the nitrogen linker, minimizing the formation of regioisomers that are common in non-directed electrophilic substitutions. Furthermore, the mild reaction temperature of 60°C helps suppress thermal decomposition pathways and polymerization of the reactive allenoate species, which can occur at higher temperatures (as evidenced by the lower yield of 57% at 100°C compared to 61% at 60°C in the patent examples). This selectivity simplifies the downstream purification process, reducing the burden on chromatography or crystallization steps and ensuring a cleaner crude profile, which is essential for meeting stringent pharmaceutical purity specifications.
How to Synthesize 2-(2-indolyl)-acetate Derivatives Efficiently
The practical execution of this synthesis is designed to be straightforward yet precise, leveraging standard laboratory equipment to achieve high reproducibility. The protocol typically involves charging the N-phenyl-2-aminopyridine substrate, the rhodium catalyst precursor, silver additives, and zinc acetate into a reaction vessel under an inert nitrogen atmosphere to prevent catalyst deactivation by oxygen. Following the addition of the solvent, 1,2-dichloroethane (DCE), and the allenoic acid ester coupling partner, the mixture is heated to maintain a steady temperature of 60°C for approximately 24 hours. This specific combination of reagents, particularly the synergistic effect of AgSbF6 and Zn(OAc)2, is critical for driving the reaction to completion while maintaining catalyst stability. 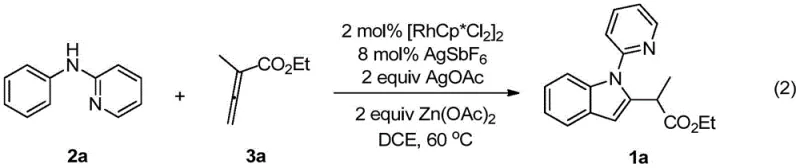 Upon completion, the workup involves simple solvent removal followed by silica gel column chromatography, demonstrating a workflow that is easily adaptable from milligram-scale discovery to multi-kilogram pilot production.
Upon completion, the workup involves simple solvent removal followed by silica gel column chromatography, demonstrating a workflow that is easily adaptable from milligram-scale discovery to multi-kilogram pilot production.
- Charge N-phenyl-2-aminopyridine, [RhCp*Cl2]2 catalyst, AgSbF6 additive, AgOAc oxidant, and Zn(OAc)2 into a reaction vessel under inert atmosphere.
- Add 1,2-dichloroethane (DCE) solvent and the allenoic acid ester coupling partner to the mixture.
- Heat the reaction mixture to 60°C for 24 hours, then purify the crude product via silica gel column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this Rh-catalyzed C-H activation technology offers tangible strategic benefits that extend beyond mere technical novelty. The primary advantage lies in the simplification of the supply chain for raw materials; since the method utilizes easily prepared N-phenyl-2-aminopyridines and commercially available allenoates, it reduces dependency on exotic or custom-synthesized building blocks that often carry long lead times and high price tags. The atom-economic nature of the reaction means that a higher proportion of the input mass is converted into the desired product, inherently reducing the volume of waste solvents and reagents that need to be disposed of, which translates to lower environmental compliance costs and a smaller carbon footprint for the manufacturing site.
- Cost Reduction in Manufacturing: The elimination of pre-functionalization steps, such as halogenation or lithiation, removes entire unit operations from the manufacturing process, leading to significant reductions in labor, energy, and consumable costs. By avoiding the use of stoichiometric amounts of expensive organometallic reagents often required in traditional cross-couplings, the process relies on a catalytic amount of rhodium, which, despite being a precious metal, is used efficiently and can potentially be recovered. The mild reaction conditions also imply lower energy consumption for heating and cooling compared to high-temperature reflux processes, contributing to a more cost-effective overall production model without compromising on yield or quality.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route against varying functional groups ensures that supply continuity is maintained even when specific substrate analogs are required. Since the core chemistry remains consistent across a wide range of R1, R2, and R3 substituents, manufacturers do not need to validate entirely new processes for every new derivative, accelerating the time-to-market for new products. The use of stable, shelf-stable reagents like silver salts and zinc acetate further mitigates the risk of supply disruptions caused by the degradation of sensitive reagents, ensuring that production schedules can be met reliably even in fluctuating market conditions.
- Scalability and Environmental Compliance: The reaction has been demonstrated to proceed effectively in standard solvents like DCE, which, while requiring careful handling, is well-understood in industrial settings with established recovery protocols. The high selectivity of the reaction minimizes the formation of difficult-to-remove byproducts, simplifying the purification train and reducing the load on wastewater treatment facilities. As regulatory pressures on pharmaceutical manufacturing increase, having a process that inherently generates less waste and avoids hazardous reagents positions the supply chain for long-term sustainability and compliance with evolving global environmental standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this indole synthesis technology. These insights are derived directly from the experimental data and scope defined in the patent literature, providing a realistic overview of what process teams can expect during technology transfer and scale-up activities.
Q: What are the critical reaction parameters for maximizing yield in this Rh-catalyzed indole synthesis?
A: According to the patent data, optimal yields (e.g., 61%) are achieved at 60°C over 24 hours using [RhCp*Cl2]2 as the catalyst precursor, AgSbF6 as an additive, and AgOAc as the oxidant in 1,2-dichloroethane. Deviating significantly from these temperatures, such as dropping to 25°C, drastically reduces conversion.
Q: Can this synthetic route accommodate diverse functional groups on the substrate?
A: Yes, the method demonstrates excellent functional group tolerance. The N-phenyl-2-aminopyridine substrate can bear various substituents including methyl, methoxy, halogens (F, Cl, Br), cyano, and acetyl groups without requiring pre-functionalization, maintaining good regioselectivity.
Q: Is the resulting indole product suitable for further downstream derivatization?
A: Absolutely. The patent explicitly demonstrates that the ester functionality on the 2-(2-indolyl)-acetate derivative can be successfully reduced to the corresponding primary alcohol using LiAlH4, proving the utility of these intermediates for generating diverse pharmacophores.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-(2-indolyl)-acetate Derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of C-H activation technologies in modernizing the synthesis of complex heterocycles. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We are committed to delivering high-purity indole derivatives that meet stringent purity specifications, utilizing our rigorous QC labs to verify every batch against the highest industry standards. By leveraging advanced catalytic methods like the one described in CN111285846B, we can offer our partners a competitive edge through superior quality and optimized manufacturing economics.
We invite you to collaborate with us to explore how this innovative synthetic route can enhance your pipeline. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific project needs. We are ready to provide specific COA data and comprehensive route feasibility assessments to help you make informed decisions about integrating these advanced intermediates into your supply chain.