Advanced Asymmetric Rearrangement for High-Purity Oxindole Intermediates with Quaternary Centers
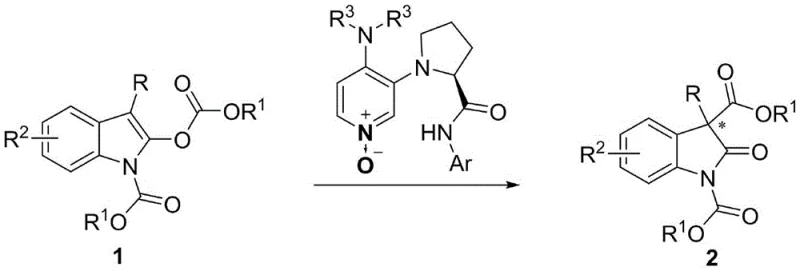
The establishment of all-carbon quaternary stereocenters remains one of the most formidable challenges in modern organic synthesis, particularly within the realm of bioactive molecule construction. Patent CN110372566B introduces a groundbreaking methodology for the asymmetric rearrangement of indole carbonates, utilizing a novel chiral 3,4-diaminopyridine nitrogen-oxygen catalyst. This technology addresses the critical need for efficient access to oxindole scaffolds containing C3 quaternary centers, which are prevalent motifs in potent anti-mitotic marine natural products and medicinal plant alkaloids. Unlike conventional approaches that often struggle with steric hindrance and low stereocontrol, this invention leverages a unique mechanistic pathway where the oxygen atom of the pyridine N-oxide acts as the nucleophilic site, distinct from the nitrogen-centered nucleophilicity of traditional DMAP catalysts. The process delivers exceptional results, achieving yields up to 98% and enantiomeric excess values reaching 96% ee under mild conditions.
For procurement managers and supply chain directors seeking a reliable pharmaceutical intermediate supplier, this technology represents a significant opportunity for cost reduction in pharmaceutical manufacturing. The reaction utilizes readily available indole carbonate precursors and operates at room temperature in common solvents like toluene, eliminating the need for cryogenic conditions or expensive transition metals. This simplicity translates directly into lower operational expenditures and reduced energy consumption during the commercial scale-up of complex pharmaceutical intermediates. Furthermore, the robustness of the catalyst system ensures consistent quality, reducing lead time for high-purity oxindole derivatives required for downstream drug development pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of oxindoles with quaternary carbon centers has relied heavily on DMAP-type organocatalysts or isothiourea derivatives. While these methods have provided some success, they suffer from inherent limitations regarding substrate scope and stereochemical control. Traditional DMAP catalysts function by utilizing the lone pair on the pyridine nitrogen to initiate nucleophilic acyl substitution. However, this mechanism often fails to provide sufficient chiral induction when bulky substituents are present at the reactive center, leading to poor enantioselectivity. Literature precedents indicate that for certain substrates, such as those with benzyl or aryl groups at the C3 position, conventional catalysts may yield products with less than 80% ee, or in some cases, fail to react entirely. Additionally, the lack of secondary interactions, such as hydrogen bonding, in these traditional systems limits the rigidity of the transition state, resulting in a broader distribution of stereoisomers that are costly and difficult to separate.
The Novel Approach
The methodology disclosed in CN110372566B fundamentally reimagines the catalytic activation mode by employing a chiral 3,4-diaminopyridine nitrogen-oxygen catalyst. In this innovative system, the nucleophilic attack is initiated by the oxygen atom of the N-oxide moiety rather than the nitrogen, creating a distinct electronic environment that favors the formation of the desired quaternary center. Crucially, the catalyst structure incorporates an amide functionality that participates in intermolecular hydrogen bonding with the substrate. This dual-activation strategy not only accelerates the reaction rate but also locks the substrate into a highly defined chiral pocket. Comparative studies within the patent demonstrate that removing the N-oxide group or methylating the amide nitrogen drastically reduces both yield and enantioselectivity, proving the synergistic effect of the oxygen nucleophile and the hydrogen bond donor. This approach successfully overcomes the steric barriers that plague older methods, enabling the efficient transformation of diverse indole carbonates into valuable chiral building blocks.
Mechanistic Insights into Chiral 3,4-Diaminopyridine Nitroxide Catalysis
To fully appreciate the value of this technology for R&D directors focused on purity and impurity profiles, a deep understanding of the catalytic cycle is essential. The reaction proceeds through a reversible nucleophilic acyl substitution between the catalyst and the indolyl carbonate substrate. This initial step generates a highly reactive O-acylated pyridinium cation intermediate and an enolate anion. The chirality is induced in the subsequent C-C bond-forming step, where the enolate attacks the acyl group of the pyridinium species. The transition state is stabilized by a critical network of non-covalent interactions, specifically the hydrogen bond between the catalyst's amide N-H proton and the carbonyl oxygen of the enolate. This interaction directs the approach of the nucleophile from a specific face of the pyridinium ring, ensuring high facial selectivity.
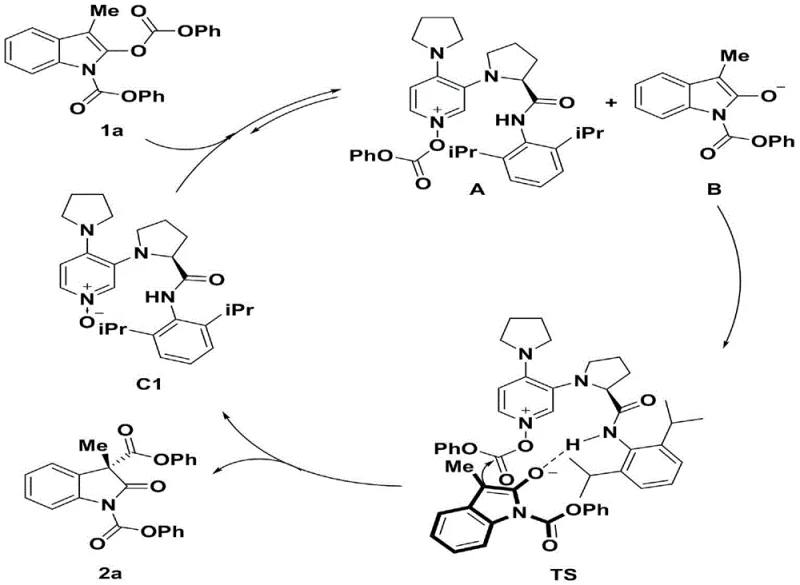
The implications for impurity control are profound. By enforcing a rigid transition state geometry through hydrogen bonding, the catalyst minimizes the formation of the undesired enantiomer and other regioisomeric byproducts. The patent data confirms that catalysts lacking the N-H proton (such as the N-methylated analog C7) result in negligible enantioselectivity (-16% ee), highlighting that the hydrogen bond is not merely auxiliary but fundamental to the stereochemical outcome. This mechanistic precision means that the crude reaction mixture contains a much higher ratio of the desired product, simplifying downstream purification. For process chemists, this translates to fewer crystallization steps or chromatographic separations, directly impacting the overall process mass intensity (PMI) and environmental footprint of the synthesis.
How to Synthesize Chiral Oxindole Derivatives Efficiently
The practical implementation of this asymmetric rearrangement is straightforward and amenable to standard laboratory and pilot plant equipment. The protocol typically involves dissolving the indole carbonate substrate in anhydrous toluene, adding the chiral catalyst C1 at a loading of 2.5 mol%, and introducing activated 3A molecular sieves to scavenge trace moisture which could deactivate the catalyst. The mixture is stirred under an inert nitrogen atmosphere at room temperature. Reaction monitoring via TLC or HPLC indicates completion typically within 36 to 48 hours, depending on the steric bulk of the substituents. Upon completion, the catalyst and molecular sieves are removed by filtration, and the product is isolated via standard aqueous workup and silica gel chromatography. The detailed standardized synthesis steps are provided in the guide below.
- Prepare the reaction mixture by combining indolyl carbonate substrate, chiral catalyst C1 (2.5 mol%), and 3A molecular sieves in anhydrous toluene under inert atmosphere.
- Stir the reaction mixture at room temperature for 36 to 48 hours to allow the asymmetric rearrangement to proceed to completion.
- Quench the reaction, perform aqueous workup, and purify the crude product via flash column chromatography to isolate the high-purity oxindole derivative.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, this catalytic technology offers compelling advantages that align with the goals of cost efficiency and supply security. The shift from precious metal catalysis or harsh stoichiometric reagents to a robust organocatalytic system eliminates the risk of heavy metal contamination, a critical compliance requirement for API manufacturing. This removal of metal catalysts significantly simplifies the purification train, reducing the consumption of specialized scavenging resins and lowering the overall cost of goods sold. Furthermore, the use of commodity solvents like toluene and the ability to run reactions at ambient temperature drastically reduce energy overheads associated with heating or cooling large reactor vessels.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven by the high turnover number of the catalyst and the elimination of expensive chiral ligands or transition metals. Since the catalyst loading is as low as 2.5 mol% and the catalyst itself is synthetically accessible from cheap starting materials, the direct material cost is minimized. Additionally, the high enantioselectivity achieved (often >90% ee) reduces the loss of material associated with recycling off-spec enantiomers, thereby maximizing the effective yield of the valuable chiral intermediate. The simplified workup procedure further contributes to cost savings by reducing labor hours and solvent usage during isolation.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the use of stable, shelf-stable reagents that do not require specialized cold-chain logistics. The starting materials, indole carbonates, are derived from widely available indoles and chloroformates, ensuring a steady upstream supply. The robustness of the reaction conditions means that the process is less sensitive to minor fluctuations in temperature or mixing rates, which enhances batch-to-batch consistency. This reliability is crucial for maintaining continuous production schedules and meeting the strict delivery timelines demanded by downstream pharmaceutical clients.
- Scalability and Environmental Compliance: The process demonstrates excellent scalability, with patent examples confirming successful gram-scale synthesis without loss of performance. The absence of toxic heavy metals and the use of benign solvents facilitate easier waste stream management and regulatory approval. The high atom economy of the rearrangement reaction, where the carbonate group serves as both the leaving group and the source of the carbonyl functionality in the product, minimizes waste generation. These factors collectively support a greener manufacturing profile, aligning with increasingly stringent global environmental regulations and corporate sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this asymmetric rearrangement technology. These answers are derived directly from the experimental data and mechanistic studies presented in the patent literature, providing a factual basis for evaluating the process viability. Understanding these details is essential for making informed decisions about adopting this methodology for your specific project needs.
Q: What is the primary advantage of the nitrogen-oxygen catalyst over traditional DMAP catalysts?
A: Unlike traditional DMAP catalysts that utilize the pyridine nitrogen as a nucleophile, this novel catalyst employs the oxygen atom of the N-oxide group as the nucleophilic site. Furthermore, the amide N-H proton plays a critical role in hydrogen bonding within the transition state, significantly enhancing enantioselectivity up to 96% ee.
Q: Can this process be scaled for commercial production of pharmaceutical intermediates?
A: Yes, the patent demonstrates successful scalability. The reaction operates under mild conditions (room temperature) with low catalyst loading (2.5 mol%), and gram-scale experiments have confirmed high yields (94%) and excellent enantiomeric excess, indicating strong potential for commercial scale-up.
Q: What types of substituents are tolerated in this asymmetric rearrangement?
A: The methodology exhibits broad substrate scope. It tolerates various groups at the C3 position including alkyl (methyl, ethyl, t-butyl), benzyl, allyl, cyano, and aryl groups. Additionally, different ester protecting groups such as phenyl, benzyl, and substituted phenyl esters are compatible with the reaction conditions.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Oxindole Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced organocatalytic methods like the one described in CN110372566B for the production of high-value pharmaceutical intermediates. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that laboratory breakthroughs are seamlessly translated into industrial reality. Our facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, including precise determination of enantiomeric excess using chiral HPLC, guaranteeing that every batch meets the exacting standards required for drug substance manufacturing.
We invite you to collaborate with us to leverage this cutting-edge chemistry for your pipeline. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific target molecule, evaluating how this rearrangement strategy can optimize your current route. Please contact our technical procurement team today to request specific COA data for similar oxindole scaffolds and to discuss route feasibility assessments for your upcoming projects.