Advanced One-Step Synthesis of Azaindole Derivatives for Scalable Pharmaceutical Intermediate Production
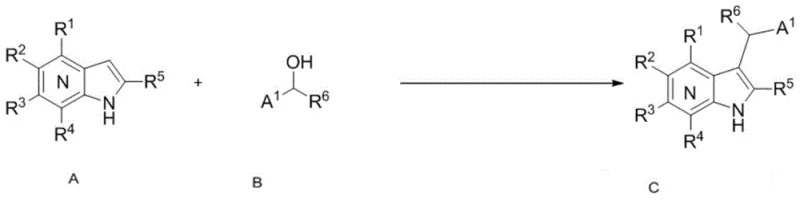
The pharmaceutical industry continuously seeks efficient pathways to construct complex heterocyclic scaffolds, particularly those exhibiting potent biological activity against malignancies. Patent CN104710417B introduces a groundbreaking methodology for the synthesis of azaindole derivatives, specifically leveraging a Friedel-Crafts alkylation strategy that transforms simple azaindole precursors and benzyl alcohols into high-value intermediates. This innovation addresses critical bottlenecks in the production of protein tyrosine kinase (PTK) inhibitors, which are pivotal in modern oncology therapeutics. By utilizing a strong acid catalyst system, the process achieves exceptional conversion rates under remarkably mild conditions, bypassing the need for elevated temperatures or prolonged reaction times that often degrade sensitive functional groups. The technical robustness of this approach positions it as a superior alternative for manufacturers aiming to secure a reliable pharmaceutical intermediates supplier capable of delivering consistent quality.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for functionalizing the azaindole core often suffer from significant inefficiencies that hinder large-scale production. Conventional methods frequently rely on multi-step sequences involving protection and deprotection strategies, which not only increase the overall material cost but also generate substantial chemical waste, complicating environmental compliance. Furthermore, many existing protocols require harsh reaction conditions, such as high temperatures or the use of toxic heavy metal catalysts, which pose safety risks and necessitate expensive purification steps to remove residual metals to meet stringent regulatory standards for active pharmaceutical ingredients. The lack of regioselectivity in older methods can also lead to complex mixtures of isomers, drastically reducing the yield of the desired C-3 substituted product and requiring resource-intensive chromatographic separations that are impractical for commercial scale-up.
The Novel Approach
In stark contrast, the methodology disclosed in CN104710417B offers a streamlined, one-pot solution that directly alkylates the azaindole ring at the C-3 position using benzyl alcohols as electrophiles. This novel approach eliminates the need for pre-activation of the alcohol substrate into halides or other leaving groups, thereby simplifying the supply chain for raw materials. The reaction proceeds efficiently at room temperature (approximately 20°C) within a short timeframe of just 2 hours, demonstrating high reaction activity and specificity. By employing trifluoromethanesulfonic acid (TfOH) as a catalyst, the process ensures rapid generation of the reactive carbocation species necessary for the electrophilic aromatic substitution, resulting in high yields without the formation of significant byproducts. This simplicity translates directly into cost reduction in pharmaceutical intermediates manufacturing by minimizing energy consumption and operational complexity.
Mechanistic Insights into TfOH-Catalyzed Friedel-Crafts Alkylation
The core of this synthetic breakthrough lies in the effective activation of the benzyl alcohol by the superacid catalyst, trifluoromethanesulfonic acid. In this mechanism, the strong acidity of TfOH facilitates the protonation of the hydroxyl group on the benzyl alcohol, promoting the loss of a water molecule to generate a stable benzylic carbocation or a tightly associated ion pair. This highly electrophilic species is then attacked by the electron-rich C-3 position of the azaindole nucleus, which acts as a nucleophile due to the resonance stabilization provided by the nitrogen atoms within the heterocyclic ring. The use of dichloromethane as a solvent is critical, as it provides a non-nucleophilic medium that stabilizes the ionic intermediates without interfering with the reaction pathway. The subsequent deprotonation restores the aromaticity of the azaindole system, yielding the final alkylated product with high fidelity.
Regioselectivity is a paramount concern in the synthesis of heterocyclic drugs, and this method excels by exclusively targeting the C-3 position. The electronic distribution of the azaindole ring makes the C-3 carbon the most nucleophilic site, ensuring that the alkylation occurs precisely where needed for biological efficacy. This specificity minimizes the formation of regioisomers that could act as impurities, thereby simplifying the impurity profile and reducing the burden on quality control laboratories. The tolerance of the reaction conditions to various substituents on both the azaindole ring and the benzyl alcohol phenyl ring further underscores its versatility. Whether dealing with electron-withdrawing halogens or electron-donating alkyl groups, the catalytic system maintains high efficiency, allowing for the rapid generation of diverse libraries of analogues for structure-activity relationship (SAR) studies.
How to Synthesize Azaindole Derivatives Efficiently
The operational simplicity of this protocol makes it highly accessible for process chemists aiming to implement this technology immediately. The procedure involves dissolving the azaindole starting material in anhydrous dichloromethane, followed by the controlled addition of the acid catalyst and the benzyl alcohol substrate. The reaction mixture is stirred at ambient temperature, monitored by thin-layer chromatography (TLC), and upon completion, undergoes a straightforward aqueous workup. Detailed standardized synthesis steps see the guide below.
- Dissolve the azaindole compound in anhydrous dichloromethane in a dried reaction flask under stirring.
- Add trifluoromethanesulfonic acid (3.0 equivalents) via syringe and stir for 2 minutes to activate the system.
- Dropwise add the benzyl alcohol substrate (3.0 equivalents), stir at room temperature for 2 hours, then neutralize and purify via silica gel chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthesis method offers tangible strategic benefits that extend beyond mere chemical efficiency. The elimination of multiple synthetic steps significantly reduces the lead time for high-purity pharmaceutical intermediates, allowing for faster response to market demands and clinical trial timelines. Since the reaction utilizes commercially available benzyl alcohols and azaindoles without requiring specialized pre-functionalized reagents, the supply chain becomes more resilient and less susceptible to disruptions caused by the scarcity of niche starting materials. Additionally, the mild reaction conditions reduce the energy footprint of the manufacturing process, aligning with corporate sustainability goals and potentially lowering utility costs associated with heating and cooling large-scale reactors.
- Cost Reduction in Manufacturing: The one-step nature of this reaction drastically cuts down on labor, solvent usage, and waste disposal costs associated with multi-step syntheses. By avoiding the use of expensive transition metal catalysts, the process removes the need for costly metal scavenging resins and extensive analytical testing for heavy metal residues, which are mandatory for API production. The high atom economy ensures that a greater proportion of raw materials ends up in the final product, maximizing the return on investment for every kilogram of input material purchased.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions means that production is less likely to be halted by minor fluctuations in environmental parameters or reagent quality. The use of common solvents like dichloromethane and standard acids ensures that raw materials are readily available from multiple global suppliers, reducing dependency on single-source vendors. This diversification of the supply base mitigates risk and ensures continuous availability of critical intermediates for downstream drug formulation.
- Scalability and Environmental Compliance: The simplicity of the post-treatment process, which involves basic neutralization and extraction, facilitates easy scale-up from pilot plants to full commercial production without the need for specialized equipment. The reduction in hazardous waste generation and the avoidance of toxic heavy metals simplify environmental permitting and compliance reporting. This green chemistry approach not only protects the environment but also enhances the company's reputation as a responsible manufacturer in the global pharmaceutical market.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this azaindole synthesis technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity for potential partners evaluating this route for their own production needs.
Q: What are the primary advantages of this azaindole synthesis method compared to traditional routes?
A: This method utilizes a direct Friedel-Crafts alkylation that proceeds in a single step under mild conditions (room temperature), eliminating the need for harsh reagents or multi-step sequences, thereby significantly reducing processing time and waste generation.
Q: Is this synthesis protocol scalable for commercial manufacturing of pharmaceutical intermediates?
A: Yes, the protocol uses common solvents like dichloromethane and standard acid catalysts, with a simple aqueous workup procedure that is highly amenable to scale-up from laboratory to industrial production volumes without complex equipment requirements.
Q: What is the regioselectivity of the alkylation on the azaindole core?
A: The reaction demonstrates high regioselectivity, specifically targeting the C-3 position of the azaindole ring system, which is crucial for maintaining the structural integrity required for downstream biological activity in kinase inhibitor development.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Azaindole Derivatives Supplier
As the demand for targeted cancer therapies continues to rise, the ability to produce high-quality azaindole intermediates efficiently is a key competitive advantage. NINGBO INNO PHARMCHEM stands ready to support your development goals with our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific requirements of this Friedel-Crafts alkylation process, ensuring that every batch meets stringent purity specifications through our rigorous QC labs. We understand the critical nature of timeline adherence in drug development and are committed to delivering materials that accelerate your progress from bench to bedside.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis route can be integrated into your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits specific to your volume requirements. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project, ensuring a partnership built on transparency, quality, and mutual success in the development of next-generation anticancer agents.