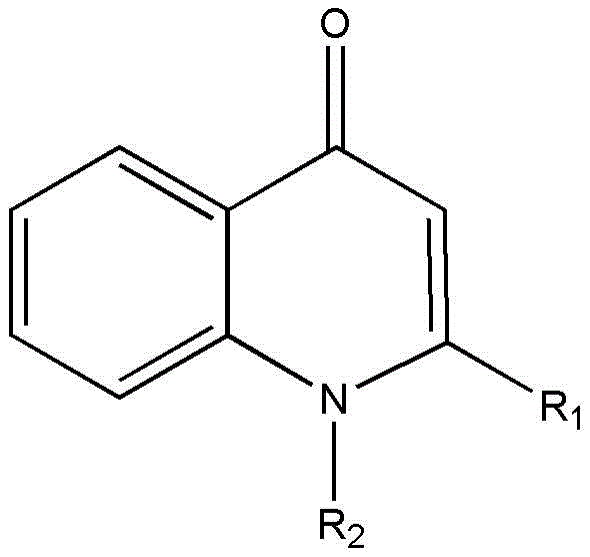
Advanced Metal-Free Synthesis of 1,2-Disubstituted-4-Quinolones for Commercial Scale-Up
Advanced Metal-Free Synthesis of 1,2-Disubstituted-4-Quinolones for Commercial Scale-Up
The pharmaceutical and fine chemical industries are constantly seeking more efficient pathways to synthesize complex heterocyclic scaffolds, particularly quinolones, which serve as critical backbones for antibacterial agents and functional materials. A significant technological breakthrough in this domain is detailed in patent CN106892866B, which introduces a novel, metal-free synthetic route for 1,2-disubstituted-4-quinolones. This innovation addresses long-standing challenges in the field by replacing expensive transition metal catalysts with a robust base-mediated cyclization strategy. For R&D directors and procurement managers alike, this shift represents a pivotal opportunity to streamline supply chains and reduce the cost of goods sold (COGS) for high-value pharmaceutical intermediates. The method leverages readily available starting materials, specifically substituted acyl chlorides and N-substituted o-aminoacetophenones, to achieve high atom utilization and exceptional purity profiles essential for downstream drug development.
Traditionally, the construction of the quinolone core has relied heavily on transition metal catalysis, often involving palladium or copper complexes that introduce significant cost and purification burdens. Conventional methods, such as those reported by Bernini et al. in 2009 or Xu Bin et al. in 2010, typically require intricate multi-step sequences to prepare specialized substrates like alkynones. These legacy processes frequently necessitate the use of hazardous reagents like n-BuLi at cryogenic temperatures (-78°C), posing severe safety risks and demanding specialized infrastructure that drives up capital expenditure. Furthermore, the reliance on noble metals like Pd(OAc)2 under high-temperature reflux conditions not only inflates raw material costs but also complicates the removal of trace metal impurities, a critical quality attribute for any reliable pharmaceutical intermediate supplier. The limitations of these older technologies create bottlenecks in both speed-to-market and overall process economics.
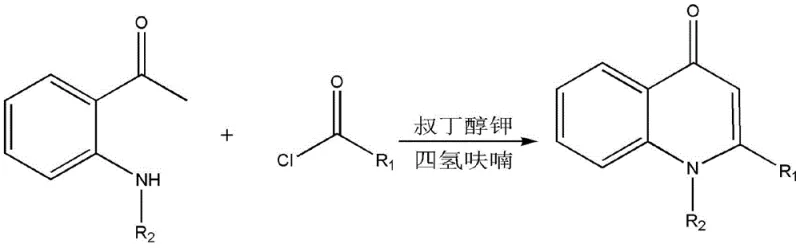
In stark contrast, the novel approach disclosed in the patent utilizes a direct condensation strategy that bypasses the need for pre-functionalized alkynes or expensive catalysts. By reacting a substituted acyl chloride directly with an N-substituted o-aminoacetophenone in the presence of a strong base, the synthesis achieves ring closure through an intramolecular Claisen-type condensation. This methodology operates under remarkably mild conditions, typically between 25°C and 60°C, eliminating the energy-intensive heating or cooling cycles associated with prior art. The general reaction scheme illustrates the simplicity and elegance of this transformation, where the structural diversity can be easily tuned by varying the R1 and R2 substituents. This flexibility allows manufacturers to access a broad library of derivatives without retooling the entire production line, offering a distinct competitive advantage in cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Base-Mediated Cyclization
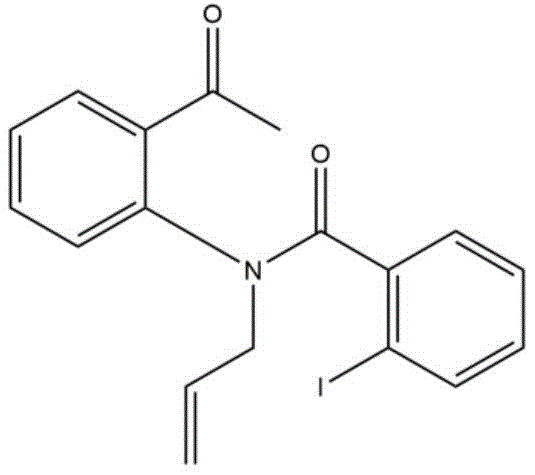
Understanding the mechanistic underpinnings of this synthesis is crucial for R&D teams aiming to optimize the process for commercial scale-up of complex quinolones. The reaction initiates with a nucleophilic attack where the lone pair of electrons on the nitrogen atom of the N-substituted o-aminoacetophenone attacks the carbonyl carbon of the substituted acyl chloride. This step forms a tetrahedral intermediate which subsequently collapses, eliminating a molecule of hydrogen chloride to generate a stable amide intermediate. This acylation step is rapid and exothermic, requiring careful temperature control during the initial mixing phase to prevent side reactions. The choice of solvent, specifically tetrahydrofuran (THF), plays a vital role in stabilizing the ionic species involved and ensuring homogeneous reaction conditions throughout the process.
Following the formation of the amide, the key cyclization event is triggered by the addition of a strong base, preferably potassium tert-butoxide (BuOK). The base abstracts the acidic alpha-proton adjacent to the ketone carbonyl group, generating a resonance-stabilized enolate or carbanion species. This nucleophilic center then attacks the amide carbonyl carbon in an intramolecular fashion, closing the six-membered ring. The final step involves the elimination of a water molecule to restore aromaticity and form the conjugated 4-quinolone system. This mechanism avoids the formation of heavy metal waste streams and ensures that the final product is free from toxic metal residues, a significant benefit for high-purity pharmaceutical intermediate applications. The structural integrity of the amide intermediate, as depicted in the patent data, confirms the stepwise nature of this transformation.
How to Synthesize 1,2-Disubstituted-4-Quinolones Efficiently
Implementing this synthesis requires precise control over stoichiometry and reaction parameters to maximize yield and minimize impurities. The process begins with the in situ or separate preparation of the acyl chloride from the corresponding carboxylic acid using thionyl chloride, followed by the N-alkylation of o-aminoacetophenone. Once these precursors are ready, they are combined in anhydrous THF under an inert atmosphere. The addition of the base must be controlled to manage the exotherm, and the reaction mixture is maintained at moderate temperatures for several hours to ensure complete conversion. Monitoring via TLC or HPLC is recommended to determine the optimal endpoint before quenching and workup. The detailed standardized synthesis steps are provided in the guide below to assist technical teams in replicating these results.
- Preparation of Substituted Acyl Chloride: React substituted carboxylic acid with thionyl chloride (SOCl2) under reflux for 1-3 hours, followed by distillation to remove excess reagent.
- Synthesis of N-Substituted o-Aminoacetophenone: React o-aminoacetophenone with an alkyl halide (e.g., allyl bromide) and potassium carbonate in DMF at 50-80°C for 22-26 hours.
- Cyclization Reaction: Mix the acyl chloride and N-substituted amine in THF, add potassium tert-butoxide (BuOK) at 25-60°C, and stir for 4-8 hours to achieve cyclization.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented technology offers transformative benefits for supply chain reliability and cost management. By eliminating the dependency on volatile and expensive noble metal catalysts like palladium and copper, manufacturers can achieve substantial cost savings in raw material procurement. The removal of these metals also simplifies the downstream purification process, as there is no need for specialized scavenging resins or complex filtration steps to meet strict residual metal specifications. This streamlining of the workflow translates directly into reduced processing time and lower operational expenditures, enhancing the overall margin profile for producers of fine chemical intermediates. Furthermore, the avoidance of hazardous reagents like n-BuLi significantly improves workplace safety and reduces the regulatory burden associated with handling pyrophoric materials.
Enhanced supply chain reliability is another critical advantage, driven by the use of commodity chemicals as starting materials. Substituted benzoic acids and aminoacetophenones are widely available from multiple global suppliers, mitigating the risk of single-source bottlenecks that often plague specialized catalyst-dependent syntheses. The mild reaction conditions (25-60°C) allow the process to be run in standard glass-lined or stainless steel reactors without the need for cryogenic cooling or high-pressure vessels, making it accessible to a broader range of contract manufacturing organizations. This accessibility fosters a more resilient supply network, ensuring consistent delivery schedules even during periods of market volatility. The robustness of the method supports reducing lead time for high-purity quinolone derivatives, enabling faster response to customer demand fluctuations.
Scalability and environmental compliance are inherently addressed by the green chemistry principles embedded in this route. The high atom utilization and absence of heavy metals result in a cleaner waste profile, simplifying effluent treatment and reducing the environmental footprint of the manufacturing site. The process has been demonstrated to achieve yields as high as 92% with specific substrates, indicating excellent efficiency and minimal waste generation. Such high efficiency is paramount for sustainable manufacturing practices and aligns with the increasing global emphasis on eco-friendly production methods. The ability to scale this reaction from laboratory benchtop to multi-ton production without significant loss in efficiency makes it an ideal candidate for long-term commercial partnerships focused on sustainable growth.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative examples provided in the patent documentation, offering clarity on substrate scope and process robustness. Understanding these nuances helps stakeholders make informed decisions about adopting this method for their specific product portfolios. The answers reflect the practical realities of running this chemistry in a production environment.
Q: Does this synthesis method require expensive transition metal catalysts?
A: No, the patented method (CN106892866B) eliminates the need for palladium or copper catalysts, utilizing a strong base like BuOK instead, which significantly reduces raw material costs and simplifies purification.
Q: What is the typical yield range for this quinolone synthesis?
A: The process demonstrates excellent efficiency with yields ranging from 72% to 92% depending on the specific substituents, with p-chlorophenyl derivatives achieving the highest conversion rates.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the reaction operates under mild conditions (25-60°C) without high-pressure equipment or cryogenic temperatures, making it highly scalable and safer for commercial manufacturing compared to traditional n-BuLi methods.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 1,2-Disubstituted-4-Quinolone Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic value of adopting advanced synthetic methodologies like the one described in CN106892866B to drive innovation in the pharmaceutical sector. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from lab-scale discovery to industrial manufacturing is seamless and efficient. We are committed to maintaining stringent purity specifications and operating rigorous QC labs to guarantee that every batch of 1,2-disubstituted-4-quinolone meets the highest international standards. Our infrastructure is designed to handle the specific requirements of base-mediated cyclizations, providing a safe and compliant environment for producing these critical intermediates.
We invite potential partners to engage with our technical procurement team to discuss how this metal-free synthesis can be integrated into your supply chain. By requesting a Customized Cost-Saving Analysis, you can quantify the potential economic benefits of switching to this greener, more efficient route. We encourage you to contact us for specific COA data and route feasibility assessments tailored to your target molecules. Together, we can leverage this cutting-edge technology to enhance product quality, reduce costs, and accelerate the delivery of life-saving medicines to the global market.