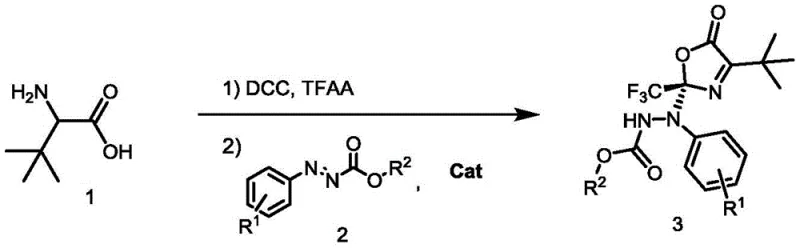
Advanced Asymmetric Synthesis of Optically Active Benzocarboxylates for High-Purity Pharmaceutical Intermediates
The pharmaceutical and fine chemical industries are constantly seeking robust methodologies for constructing quaternary carbon stereocenters, a structural motif prevalent in numerous bioactive molecules. Patent CN113200933A introduces a groundbreaking approach for the synthesis of optically active benzocarboxylate compounds through an asymmetric addition reaction. This technology leverages tert-leucine as a cost-effective starting material, which is initially converted into a 2-trifluoroalkyl oxazol-5(2H)-one intermediate. Subsequently, in the presence of a sophisticated chiral bifunctional tertiary amine urea catalyst, this intermediate undergoes a highly stereoselective addition with azocarboxylic esters. The significance of this invention lies in its ability to achieve high optical purity and yield in a one-pot, two-step sequence, addressing critical challenges in the manufacturing of complex chiral building blocks.
For R&D directors and process chemists, the introduction of perfluoroalkyl functional groups into chiral scaffolds is of paramount importance due to their profound impact on the metabolic stability and lipophilicity of drug candidates. The disclosed method not only facilitates the formation of carbon-nitrogen bonds at the sterically hindered C-2 position of the azlactone ring but does so with exceptional control over stereochemistry. By utilizing a metal-free organocatalytic system, the process circumvents the stringent regulatory hurdles associated with heavy metal residues in active pharmaceutical ingredients (APIs). This represents a significant leap forward in green chemistry principles applied to high-value intermediate synthesis.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of nitroxylamino derivatives containing quaternary carbon chiral centers has been fraught with difficulties. Conventional strategies often rely on transition metal catalysis, which necessitates expensive ligands and rigorous exclusion of air and moisture, thereby inflating production costs and complicating scale-up operations. Furthermore, traditional multi-step syntheses typically require the isolation of unstable azlactone intermediates, leading to significant material loss and increased solvent consumption. The formation of carbon-nitrogen bonds at the C-2 position of azlactones with high enantioselectivity has remained a persistent challenge, with many existing methods suffering from poor substrate scope or requiring cryogenic temperatures that are energy-intensive and impractical for large-scale manufacturing.
The Novel Approach
The methodology described in patent CN113200933A offers a transformative solution by employing a one-pot tandem reaction strategy. Instead of isolating the reactive 2-trifluoroalkyl oxazol-5(2H)-one, it is generated in situ using dicyclohexylcarbodiimide (DCC) and trifluoroacetic anhydride (TFAA) in mesitylene. This intermediate is then immediately subjected to asymmetric addition with azocarboxylic esters catalyzed by a chiral urea derivative. This approach drastically simplifies the operational workflow, eliminating unit operations associated with intermediate isolation and purification. The use of mild reaction conditions, specifically at 25°C under an oxygen atmosphere, enhances process safety and reduces energy expenditure, making it an economically superior alternative for the commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Chiral Urea-Catalyzed Asymmetric Addition
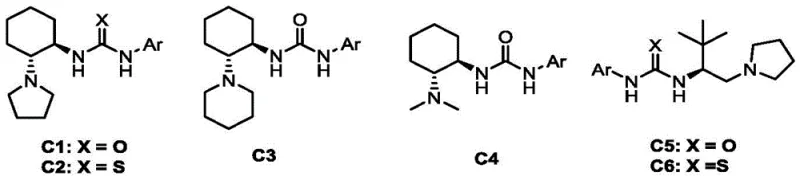
The core of this technological breakthrough resides in the unique activation mode provided by the chiral bifunctional tertiary amine urea catalysts. As illustrated in the catalyst library, structures such as C3, derived from 1R,2R-cyclohexane-1,2-diamine, play a pivotal role in orchestrating the stereochemical outcome of the reaction. These catalysts operate through a dual-activation mechanism wherein the urea moiety forms a hydrogen-bonding network with the electrophilic azlactone intermediate, thereby increasing its reactivity towards nucleophilic attack. Simultaneously, the tertiary amine component may assist in deprotonating or organizing the nucleophile, creating a highly ordered chiral environment that directs the facial selectivity of the addition.
This precise molecular recognition ensures that the nucleophilic attack occurs exclusively from one face of the planar azlactone intermediate, resulting in the formation of a single enantiomer with high fidelity. The presence of the trifluoromethyl group on the oxazole ring further modulates the electronic properties of the substrate, enhancing its electrophilicity while the bulky tert-butyl group provides steric differentiation. From an impurity control perspective, the high specificity of the organocatalyst minimizes the formation of diastereomers and regioisomers, which are common byproducts in non-catalyzed or metal-catalyzed variants. This inherent selectivity translates directly to a cleaner crude reaction profile, significantly reducing the burden on downstream purification processes and ensuring the delivery of high-purity pharmaceutical intermediates that meet stringent quality specifications.
How to Synthesize Optically Active Benzocarboxylate Efficiently
The synthesis protocol outlined in the patent provides a clear roadmap for replicating these high-performance results in a laboratory or pilot plant setting. The process begins with the activation of tert-leucine in an anhydrous solvent, followed by the sequential addition of reagents to generate the key azlactone species. Once the intermediate is formed, the introduction of the azo ester and the specific catalyst C3 initiates the stereoselective coupling. The detailed standardized synthesis steps below outline the precise stoichiometry, temperature controls, and workup procedures required to achieve the reported yields and enantiomeric excess values.
- Generate the 2-trifluoroalkyl oxazol-5(2H)-one intermediate by reacting tert-leucine with trifluoroacetic anhydride and DCC in mesitylene at 25°C.
- Add the azocarboxylic ester and chiral bifunctional tertiary amine urea catalyst (e.g., C3) to the reaction mixture under oxygen atmosphere.
- Stir the reaction at room temperature for 12 hours, followed by filtration, solvent removal, and silica gel column chromatography purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers compelling economic and logistical benefits. The shift from precious metal catalysis to organocatalysis fundamentally alters the cost structure of the manufacturing process. By eliminating the need for expensive palladium, rhodium, or iridium complexes, the raw material costs are significantly reduced. Moreover, the absence of heavy metals removes the necessity for specialized scavenging resins and extensive analytical testing for metal residues, which are costly and time-consuming steps in API production. This streamlined approach facilitates cost reduction in pharmaceutical intermediate manufacturing by simplifying the supply chain and reducing the dependency on volatile precious metal markets.
- Cost Reduction in Manufacturing: The utilization of readily available starting materials like tert-leucine and commodity reagents such as DCC and TFAA ensures a stable and low-cost input stream. The one-pot nature of the reaction minimizes solvent usage and waste disposal costs, as there is no need for intermediate isolation. Furthermore, the recyclability and reusability of the chiral urea catalyst, as noted in the patent benefits, contribute to long-term operational savings. The mild reaction conditions also reduce energy consumption for heating or cooling, further driving down the overall cost of goods sold (COGS) without compromising on quality or yield.
- Enhanced Supply Chain Reliability: Sourcing complex chiral catalysts can often be a bottleneck; however, the catalysts described herein are derived from abundant chiral diamines and can be synthesized in-house or sourced reliably. The robustness of the reaction conditions, which tolerate oxygen and proceed at ambient temperature, reduces the risk of batch failures due to equipment malfunction or environmental fluctuations. This reliability ensures consistent production schedules and shorter lead times, allowing supply chain managers to maintain optimal inventory levels and respond swiftly to market demands for reliable pharmaceutical intermediate suppliers.
- Scalability and Environmental Compliance: The process is inherently scalable due to its simplicity and the use of standard organic solvents like mesitylene. The reduction in step count and the avoidance of hazardous heavy metals align perfectly with modern environmental, health, and safety (EHS) regulations. This compliance reduces the regulatory burden and potential liabilities associated with chemical manufacturing. The high atom economy and reduced waste generation support sustainability goals, making this technology attractive for companies aiming to minimize their environmental footprint while scaling up production capacities for global distribution.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this asymmetric synthesis technology. These answers are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing clarity on the practical application and advantages of this method for industrial partners.
Q: What is the primary advantage of using chiral urea catalysts in this synthesis?
A: Chiral bifunctional tertiary amine urea catalysts, such as C3 derived from 1R,2R-cyclohexane-1,2-diamine, provide excellent enantioselectivity (up to 96% ee) and high yields without requiring toxic transition metals, simplifying downstream purification.
Q: Can this process be scaled for commercial production of API intermediates?
A: Yes, the process utilizes readily available starting materials like tert-leucine and operates under mild conditions (25°C, atmospheric pressure), making it highly suitable for commercial scale-up with reduced safety risks and operational costs.
Q: How does the one-pot two-step method impact production efficiency?
A: By generating the reactive azlactone intermediate in situ and immediately subjecting it to asymmetric addition, the method eliminates the need for isolating unstable intermediates, significantly reducing solvent usage, waste generation, and overall processing time.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Optically Active Benzocarboxylate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthetic routes in the development of next-generation therapeutics. Our team of expert process chemists has extensively evaluated the technology disclosed in CN113200933A and confirmed its potential for industrial application. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to full-scale manufacturing is seamless. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications and enantiomeric excess, guaranteeing that every batch of optically active benzocarboxylate meets the highest industry standards.
We invite pharmaceutical companies and research institutions to collaborate with us to optimize this synthesis for their specific pipeline needs. By leveraging our expertise in organocatalysis and process intensification, we can help you achieve substantial efficiencies in your supply chain. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your project. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how partnering with us can accelerate your drug development timeline and reduce overall production costs.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →