Advanced Cyclization Technology for High-Purity Entecavir Intermediates and Commercial Scalability
Advanced Cyclization Technology for High-Purity Entecavir Intermediates and Commercial Scalability
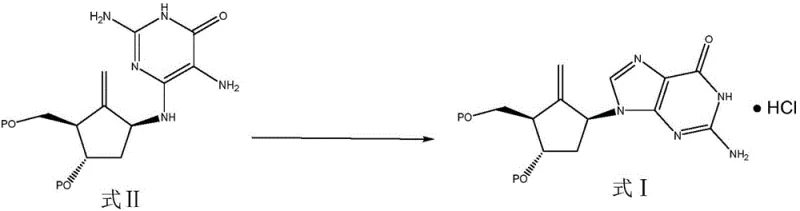
The global demand for effective antiviral therapeutics continues to drive innovation in the synthesis of complex nucleoside analogues, particularly for the treatment of chronic hepatitis B. Entecavir stands as a cornerstone in this therapeutic landscape, yet the economic and technical viability of its production often hinges on the efficiency of its key intermediate synthesis. Patent CN110759912A introduces a transformative preparation method that addresses long-standing bottlenecks in the manufacturing of the entecavir intermediate, specifically the conversion of the open-chain precursor (Formula II) into the cyclized core structure (Formula I). This technical breakthrough leverages a sophisticated acid-catalyzed ring-closure reaction in the presence of trialkyl orthoformate, achieving conversion rates that significantly surpass historical benchmarks. For R&D directors and procurement strategists, this patent represents not merely a chemical adjustment but a fundamental shift towards a more robust, high-yield, and commercially viable supply chain for high-purity pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the innovations detailed in CN110759912A, the industry relied heavily on methodologies such as those disclosed in CN200610088464.8, which suffered from inherent inefficiencies that plagued large-scale operations. The conventional approach was characterized by a problematic reaction profile where the conversion of the starting material to the desired product was incomplete, leaving significant amounts of unreacted intermediate states in the reaction mixture. This lack of complete conversion necessitated complex and costly post-treatment procedures, including neutralization and ion exchange resin processing, to isolate the off-white solid product. Furthermore, the reliance on external standard methods to measure product content often revealed yields far below expectations, creating a volatile supply environment where batch-to-batch consistency was difficult to guarantee. The difficulty in purifying the crude product from these incomplete reactions directly impacted the overall cost of goods sold and introduced potential impurity risks that are unacceptable in modern GMP manufacturing environments.
The Novel Approach
In stark contrast, the novel approach described in the patent utilizes a streamlined cyclization strategy that maximizes atomic economy and reaction throughput. By employing a strong acid catalyst, specifically concentrated hydrochloric acid, in conjunction with trialkyl orthoformate and a carefully selected organic solvent, the reaction drives the equilibrium decisively towards the formation of the cyclic Formula I compound. This method effectively minimizes the accumulation of intermediate states that previously hampered yield, resulting in a much cleaner reaction profile. The process operates under moderate thermal conditions, typically ranging from 45°C to 55°C, which reduces energy consumption compared to high-temperature alternatives. Moreover, the workup procedure is drastically simplified; instead of complex resin treatments, the product can be isolated through a straightforward cooling and crystallization process at -5°C, followed by filtration. This operational simplicity translates directly into reduced processing time and lower operational expenditures for manufacturers.
Mechanistic Insights into Acid-Catalyzed Cyclization
The core of this technological advancement lies in the precise manipulation of reaction kinetics through acid catalysis and solvent engineering. The mechanism involves the activation of the amino group on the pyrimidine ring of the Formula II precursor by the strong acid, facilitating a nucleophilic attack that closes the ring to form the imidazole moiety found in Formula I. The presence of trialkyl orthoformate serves a dual purpose: it acts as a dehydrating agent to drive the equilibrium forward by removing water generated during the cyclization, and it stabilizes the transition state. Crucially, the choice of solvent plays a pivotal role in stabilizing the charged intermediates and ensuring homogeneity throughout the reaction vessel. Experimental data within the patent demonstrates that polar aprotic solvents like DMF and acetonitrile provide superior solvation properties compared to protic solvents like methanol or non-polar solvents like dichloromethane, leading to markedly higher conversion efficiencies. This mechanistic understanding allows process chemists to fine-tune the reaction parameters to suppress side reactions and maximize the formation of the desired stereochemistry.
Impurity control is another critical aspect where this mechanism excels, addressing the concerns of R&D directors regarding product quality. In previous methods, the persistence of intermediate states led to a complex impurity profile that was difficult to separate from the final API. The new method's ability to push conversion rates above 75% inherently reduces the load of starting material-related impurities. Furthermore, the crystallization step at low temperatures (-5°C) acts as a powerful purification tool, leveraging the differential solubility of the product versus potential byproducts. By controlling the cooling rate and stirring duration during crystallization, manufacturers can ensure that the resulting filter cake possesses high purity without the need for additional chromatographic steps. This intrinsic purity build-up during synthesis is a hallmark of a well-designed process, reducing the burden on downstream purification units and ensuring a consistent quality profile suitable for regulatory submission.
How to Synthesize Entecavir Intermediate Efficiently
Implementing this synthesis route requires strict adherence to the optimized parameters defined in the patent to ensure reproducibility and safety. The process begins with the preparation of a diluted hydrochloric acid solution, which is then added to a mixture of the Formula IIa precursor, triethyl orthoformate, and the chosen solvent under controlled agitation. Maintaining the feed liquid temperature around 25°C during the acid addition is critical to prevent exothermic runaway and ensure uniform mixing. Following the addition, the reaction mixture is heated to the optimal range of 53°C ± 1°C and maintained for an extended period, typically exceeding 30 hours, to allow the slow cyclization to reach completion. Monitoring via HPLC is essential to determine the exact endpoint, ensuring that the conversion has maximized before proceeding to the isolation phase. The detailed standardized synthetic steps for this high-efficiency route are outlined in the guide below.
- Prepare the reaction mixture by dissolving the Formula II precursor in a suitable solvent such as acetonitrile or DMF, along with triethyl orthoformate.
- Add a hydrochloric acid solution under controlled temperature conditions (around 25°C) and stir to initiate the ring-closing reaction.
- Heat the mixture to 45-55°C for extended reaction time, then cool to -5°C to induce crystallization and filter to collect the high-purity product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented methodology offers substantial strategic benefits that extend beyond simple chemical yield. The shift from low-efficiency processes to this high-conversion protocol fundamentally alters the cost structure of entecavir intermediate manufacturing. By eliminating the need for expensive and time-consuming ion exchange resin treatments and complex neutralization steps, the process significantly reduces the consumption of auxiliary materials and labor hours. The higher conversion rate means that less raw material is wasted per kilogram of finished product, directly lowering the variable cost of production. Additionally, the use of common, industrially available solvents like acetonitrile and DMF ensures that the supply chain remains resilient against raw material shortages, unlike processes that rely on exotic or highly specialized reagents. This reliability is crucial for maintaining continuous production schedules and meeting the rigorous delivery timelines demanded by global pharmaceutical clients.
- Cost Reduction in Manufacturing: The elimination of complex post-treatment steps such as ion exchange resin processing leads to a drastic simplification of the workflow, thereby reducing operational costs associated with equipment usage and waste disposal. The higher yield per batch means that the fixed costs of production, including energy and facility overhead, are amortized over a larger quantity of saleable product, enhancing overall margin potential. Furthermore, the simplified crystallization workup reduces the volume of solvent required for washing and purification, contributing to lower utility costs and a smaller environmental footprint.
- Enhanced Supply Chain Reliability: The robustness of this reaction against variations in conditions ensures consistent batch-to-batch quality, which is vital for long-term supply contracts. The use of stable, non-hazardous reagents like triethyl orthoformate and hydrochloric acid minimizes the risks associated with the storage and transport of dangerous chemicals, streamlining logistics and compliance. This stability allows manufacturers to hold strategic stockpiles of key intermediates with confidence, knowing that the shelf-life and reactivity are predictable, thus buffering the supply chain against market volatility.
- Scalability and Environmental Compliance: The process is inherently scalable, moving seamlessly from laboratory benchtop to multi-ton industrial reactors without the need for significant re-engineering of the reaction parameters. The reduction in waste generation, particularly the avoidance of salt-heavy waste streams from neutralization steps, aligns with increasingly stringent environmental regulations and sustainability goals. This green chemistry advantage not only mitigates regulatory risk but also enhances the brand reputation of the supplier as a responsible partner in the pharmaceutical value chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this entecavir intermediate synthesis technology. These insights are derived directly from the experimental data and technical specifications provided in patent CN110759912A, offering clarity on reaction conditions, solvent selection, and scalability. Understanding these details is essential for technical teams evaluating the feasibility of integrating this route into their existing manufacturing capabilities. The answers provided reflect the consensus of current chemical engineering best practices applied to this specific nucleoside analogue synthesis.
Q: What represents the primary improvement of this new synthesis method over prior art?
A: The primary improvement is the significant increase in reaction conversion rate, reaching over 75%, compared to the lower yields and difficult post-treatment associated with previous methods like CN200610088464.8.
Q: Which solvents provide the optimal conversion rates for this cyclization reaction?
A: Experimental data indicates that polar aprotic solvents such as DMF and acetonitrile yield the highest conversion rates (84.5% and 79.4% respectively), outperforming solvents like acetone or dichloromethane.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the method is explicitly designed for industrial scalability, utilizing common reagents like triethyl orthoformate and standard crystallization techniques that facilitate easy filtration and purification.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Entecavir Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent literature to commercial reality requires a partner with deep technical expertise and proven manufacturing capacity. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the promising yields described in CN110759912A can be realized in a GMP-compliant environment. We are committed to delivering high-purity entecavir intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our facility is designed to handle the specific solvent systems and thermal requirements of this cyclization process, guaranteeing a supply continuity that supports your drug development and commercialization timelines.
We invite you to engage with our technical procurement team to discuss how this advanced synthesis route can optimize your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic benefits specific to your volume requirements. We encourage potential partners to contact us directly to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions based on hard data and our demonstrated capability to deliver excellence in pharmaceutical intermediate manufacturing.