Advanced Manufacturing of Sacubitril Intermediate N-Boc Amino Alcohol for Global Pharma Supply Chains
The pharmaceutical landscape for cardiovascular therapeutics continues to evolve, with Sacubitril (AHU-377) standing as a cornerstone component of the groundbreaking heart failure medication LCZ696. As detailed in the recent patent CN114957043A, published on August 30, 2022, there has been a significant technological breakthrough in the preparation of the critical chiral intermediate, N-Boc amino alcohol [formula (10-a)]. This intermediate serves as the structural backbone for neutral endopeptidase (NEP) inhibitors, dictating the efficacy and safety profile of the final active pharmaceutical ingredient (API). For global supply chain stakeholders, understanding this novel synthetic pathway is not merely an academic exercise but a strategic imperative for securing reliable sources of high-purity pharmaceutical intermediates. The disclosed method addresses long-standing bottlenecks in traditional synthesis, offering a route that is economically viable, environmentally friendlier, and robust enough for multi-ton commercial production.
The urgency for optimized manufacturing processes stems from the complex molecular architecture of Sacubitril, which requires precise stereochemical control to ensure therapeutic effectiveness. The patent highlights that while previous methods existed, they were fraught with inefficiencies that hampered cost-effective scale-up. By leveraging a cascade of reactions starting from readily available commodity chemicals, this new technology promises to redefine the cost structure and supply reliability for this vital cardiac medication. For procurement managers and R&D directors alike, the shift towards this methodology represents a tangible opportunity to enhance margin stability and reduce dependency on scarce, high-cost reagents that have historically plagued the supply chain.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of the key N-Boc amino alcohol intermediate has relied on pathways that are inherently inefficient and costly for large-scale industrial application. Prominent prior art, such as the route described in J. Med. Chem. 1995, utilizes D-tyrosine as a starting material. While chemically feasible, D-tyrosine is an unnatural amino acid that commands a premium price in the fine chemical market, directly inflating the cost of goods sold (COGS). Furthermore, this legacy process necessitates the use of trifluoromethanesulfonic anhydride, a reagent known for its extreme reactivity and corrosiveness. Handling such hazardous materials requires specialized, corrosion-resistant reactor equipment and stringent safety protocols, which adds substantial capital expenditure and operational complexity to the manufacturing facility.
Alternative routes disclosed in patents like WO2014032627 and CN 105026361 attempt to bypass the tyrosine route but introduce their own set of severe drawbacks. These methods often rely on stoichiometric amounts of triphenylphosphine and azodicarboxylates (Mitsunobu-type conditions). The generation of large quantities of triphenylphosphine oxide as a byproduct creates a significant purification burden, requiring extensive chromatography or crystallization steps to remove, which drastically reduces overall yield. Moreover, azodicarboxylates are thermally unstable and shock-sensitive, posing potential safety hazards during heating processes on a large scale. Other enzymatic approaches, while green in theory, suffer from long synthetic sequences and the high cost of specialized biocatalysts and cofactors like NADP+, making them less competitive for bulk manufacturing compared to robust chemical synthesis.
The Novel Approach
In stark contrast to these cumbersome legacy methods, the process outlined in patent CN114957043A introduces a streamlined, convergent synthesis that fundamentally alters the economic equation. The new route begins with inexpensive, commodity-grade raw materials such as benzaldehyde, ammonia, and (S)-epichlorohydrin. This shift away from specialty chiral pools to basic petrochemical derivatives results in a drastic reduction in raw material costs. The core innovation lies in the construction of the chiral aziridine intermediate, which serves as a versatile electrophile for the subsequent carbon-carbon bond-forming step. By eliminating the need for precious metal catalysts like Rhodium or Palladium in the early stages and avoiding hazardous sulfonic anhydrides, the process significantly lowers the barrier to entry for safe, large-scale production.
Furthermore, the novel approach is designed with process intensification in mind. The patent explicitly describes the capability for continuous operation and one-pot transformations, particularly in the later stages where cyclization, hydrolysis, and protection can be telescoped. This minimizes the number of isolation steps, reduces solvent consumption, and lowers energy requirements associated with drying and heating multiple intermediates. For a reliable pharmaceutical intermediates supplier, this translates to a more agile production schedule and a reduced environmental footprint, aligning perfectly with modern green chemistry principles and regulatory expectations for sustainable manufacturing practices.
Mechanistic Insights into Copper-Catalyzed Aziridine Ring Opening
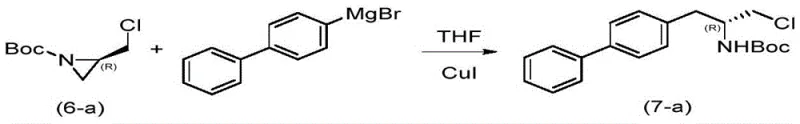
The heart of this synthetic strategy is the copper-catalyzed nucleophilic ring-opening of the chiral aziridine intermediate (compound 6) with a biphenyl Grignard reagent (compound M). This transformation is critical as it establishes the biphenyl scaffold essential for the biological activity of Sacubitril while simultaneously setting the stereocenter. The mechanism involves the coordination of the cuprous salt catalyst (such as CuI) to the nitrogen atom of the aziridine ring, which activates the ring towards nucleophilic attack. The Grignard reagent then attacks the less hindered carbon of the aziridine ring in an SN2-like fashion. Crucially, the patent data indicates that starting from (S)-epichlorohydrin leads to an (R)-configured aziridine, which upon ring opening retains the desired stereochemistry for the final API. This high level of stereocontrol is paramount for R&D directors concerned with impurity profiles and enantiomeric excess (ee) specifications.
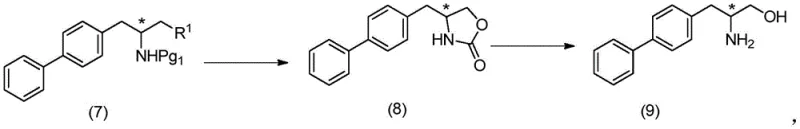
Following the coupling reaction, the resulting chloro-amine intermediate (compound 7) undergoes a fascinating intramolecular cyclization to form an oxazolidinone ring (compound 8). This cyclization serves a dual purpose: it protects the amine functionality temporarily and activates the adjacent carbon for the subsequent hydrolysis step. The hydrolysis under basic conditions cleaves the oxazolidinone ring to reveal the free amino alcohol (compound 9), which is then immediately protected with a Boc group to yield the stable final intermediate (10-a). This sequence of cyclization and hydrolysis is mechanistically elegant because it avoids the need for separate protection and deprotection steps that would otherwise add length and cost to the synthesis. The ability to perform these transformations in a telescoped manner, as shown in the reaction scheme below, demonstrates a deep understanding of physical organic chemistry applied to process optimization.
From an impurity control perspective, this mechanism offers distinct advantages. The use of catalytic copper (as low as 0.01 eq) minimizes the residual heavy metal load in the final product, simplifying the purification workflow. Traditional routes often struggle with removing stoichiometric metal waste, which can require expensive scavenging resins. Here, the workup involves simple aqueous quenching and extraction, which is highly scalable. The formation of the oxazolidinone intermediate also acts as a purification checkpoint; impurities that do not cyclize can be easily separated before the final hydrolysis step. This inherent self-purifying nature of the reaction pathway ensures that the final high-purity pharmaceutical intermediates meet the stringent quality standards required for regulatory filing, reducing the risk of batch rejection and supply disruption.
How to Synthesize Sacubitril Intermediate N-Boc Amino Alcohol Efficiently
The practical implementation of this synthesis requires careful attention to reaction conditions, particularly regarding temperature control and moisture exclusion during the Grignard coupling step. The patent provides detailed embodiments ranging from gram-scale optimization to kilogram-scale validation, offering a clear roadmap for technology transfer. The process begins with the preparation of the chiral aziridine building block, followed by the critical coupling reaction in tetrahydrofuran (THF) at low temperatures (-15°C to -20°C) to prevent side reactions. Subsequent steps involve heating in toluene to effect cyclization and hydrolysis. The robustness of this method is evidenced by its successful demonstration in Example 7, where over 1 kg of starting material was processed to yield the final product with excellent purity (99.3%) and yield. For detailed standard operating procedures and specific parameter settings, please refer to the guide below.
- Preparation of chiral aziridine intermediate (6-a) from benzaldehyde and (S)-epichlorohydrin via mesylation and intramolecular cyclization.
- Copper-catalyzed ring-opening coupling of aziridine (6-a) with biphenylmagnesium bromide to form the biphenyl backbone (7-a).
- One-pot cyclization to oxazolidinone (8-a), followed by alkaline hydrolysis and Boc-protection to yield the final N-Boc amino alcohol (10-a).
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthesis route offers compelling strategic advantages that extend beyond simple chemistry. The most significant benefit is the radical simplification of the raw material supply base. By replacing expensive, niche chiral starters like D-tyrosine with ubiquitous commodities like benzaldehyde and epichlorohydrin, manufacturers can insulate themselves from the volatility of the specialty chemical market. This shift ensures a more stable and predictable supply chain, reducing the risk of production stoppages due to raw material shortages. Additionally, the elimination of hazardous reagents like triflic anhydride reduces the regulatory burden and safety compliance costs associated with handling and storing dangerous goods, further contributing to overall operational efficiency.
- Cost Reduction in Manufacturing: The economic impact of this process is profound, driven primarily by the substitution of high-cost reagents with low-cost alternatives. The avoidance of precious metal catalysts (Rh, Pd) and stoichiometric phosphine reagents eliminates significant line items from the bill of materials. Furthermore, the ability to telescope multiple reaction steps into one-pot operations drastically reduces solvent usage, energy consumption for heating and cooling, and labor hours required for intermediate isolations. These cumulative efficiencies translate into a substantially lower cost of goods sold, allowing for more competitive pricing in the generic API market while maintaining healthy margins for the manufacturer.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the simplicity and robustness of the reaction conditions. The process utilizes standard laboratory and plant equipment without the need for exotic, corrosion-lined reactors required for strong acids. The reaction times are relatively short, and the workup procedures involve standard aqueous extractions and crystallizations, which are easily scalable from pilot plant to commercial production. This ease of scale-up means that suppliers can respond more rapidly to fluctuations in demand, ensuring consistent delivery schedules for downstream API manufacturers and mitigating the risk of drug shortages in the cardiovascular therapeutic sector.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this route is markedly superior. The reduction in solid waste (such as triphenylphosphine oxide) and the minimization of solvent exchanges align with green chemistry metrics. The process generates fewer hazardous byproducts, simplifying waste treatment and disposal protocols. This not only reduces environmental compliance costs but also enhances the sustainability profile of the supply chain, a factor that is increasingly important for multinational pharmaceutical companies aiming to meet their corporate social responsibility (CSR) goals. The demonstrated kilogram-scale success in the patent confirms that these environmental benefits are achievable at commercial volumes.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. They are derived from the specific experimental data and advantageous effects described in the patent documentation, providing clarity on scalability, purity, and operational feasibility. Understanding these details is crucial for technical teams evaluating the viability of integrating this route into their existing manufacturing portfolios.
Q: What are the key advantages of this new synthesis route over traditional D-Tyrosine methods?
A: The new route avoids expensive unnatural amino acids and corrosive triflic anhydride, utilizing cheap commodity chemicals like benzaldehyde and epichlorohydrin, significantly lowering raw material costs and equipment corrosion risks.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the patent demonstrates kilogram-scale examples with one-pot operations that minimize solvent swaps and isolation steps, proving high scalability and reduced energy consumption for commercial manufacturing.
Q: How is stereochemical purity controlled in this synthesis?
A: Stereocontrol is achieved starting from chiral (S)-epichlorohydrin, which undergoes inversion during aziridine formation and subsequent coupling, ensuring the final product maintains the required (R)-configuration with high optical purity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Sacubitril Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition to a more efficient synthesis route requires a partner with deep technical expertise and proven manufacturing capabilities. As a leading CDMO, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this patent are fully realized in practice. Our state-of-the-art facilities are equipped to handle the specific requirements of this chemistry, including low-temperature cryogenic reactions and rigorous moisture control, guaranteeing consistent quality. We adhere to stringent purity specifications and operate rigorous QC labs to ensure that every batch of Sacubitril intermediate meets the highest international standards for safety and efficacy.
We invite you to collaborate with us to optimize your supply chain for this critical cardiovascular medication. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how this new route can improve your bottom line. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. By partnering with us, you secure not just a chemical supplier, but a strategic ally committed to driving innovation and efficiency in the global pharmaceutical supply chain.