Scalable Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles for Advanced Pharmaceutical Applications
The pharmaceutical and agrochemical industries continuously seek robust synthetic routes for nitrogen-containing heterocycles, particularly those incorporating fluorine motifs which are renowned for enhancing metabolic stability and bioavailability. Patent CN110467579B introduces a significant advancement in this domain by disclosing a preparation method for 5-trifluoromethyl substituted 1,2,4-triazole compounds. This technology addresses critical bottlenecks in the synthesis of these valuable scaffolds, which are ubiquitous in high-value active pharmaceutical ingredients (APIs) such as antifungal agents and kinase inhibitors. The disclosed methodology leverages a non-metallic iodine promotion strategy, circumventing the regulatory and purification hurdles associated with transition metal catalysis. By utilizing inexpensive and commercially accessible starting materials like hydrazones and trifluoroethylimidoyl chlorides, this invention offers a streamlined pathway that is both economically viable and operationally simple.
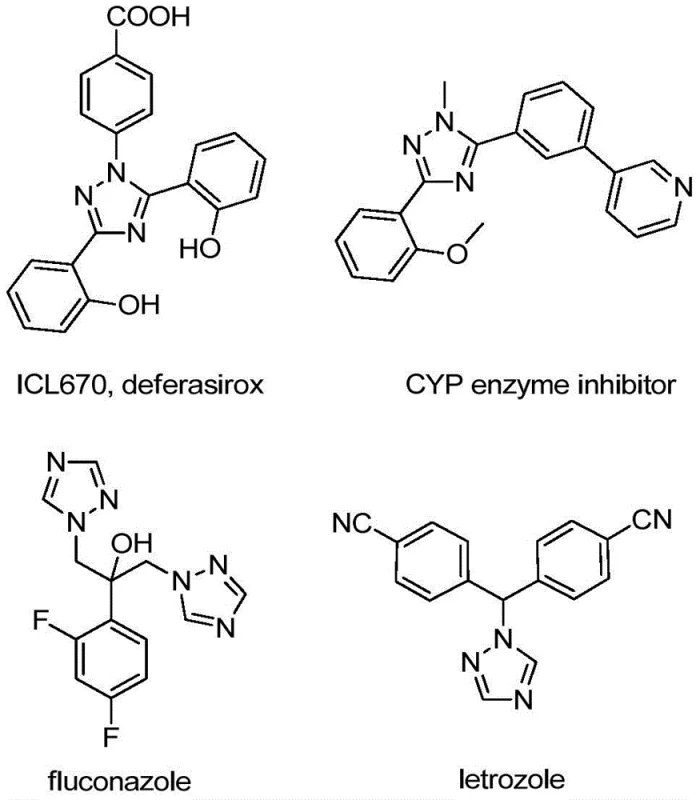
The structural versatility of the 1,2,4-triazole ring is evident in its presence across a wide spectrum of therapeutic classes, ranging from iron chelators to aromatase inhibitors. The introduction of a trifluoromethyl group at the 5-position further amplifies the pharmacological potential of these molecules by modulating their electronic properties and lipophilicity. For R&D directors and process chemists, the ability to access these substituted heterocycles through a reliable, scalable, and safe protocol is paramount. The patent data suggests that this new route not only simplifies the synthetic sequence but also broadens the applicability of the method to variously substituted substrates, thereby serving as a powerful tool for medicinal chemistry campaigns aiming to optimize lead compounds.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of trifluoromethyl-substituted nitrogen heterocycles has been fraught with challenges that impede efficient manufacturing. Traditional strategies often rely on the direct trifluoromethylation of pre-synthesized heterocyclic cores, a process that frequently necessitates the use of specialized, costly, and sometimes hazardous trifluoromethylating reagents. Alternatively, methods employing trifluorodiazoethane as a synthon have been reported; however, the inherent instability and explosive nature of diazo compounds pose severe safety risks, particularly when attempting to scale reactions from milligram to kilogram quantities. Furthermore, many existing protocols depend heavily on transition metal catalysts, such as copper or palladium complexes, which introduce significant complications regarding metal residue removal. For a reliable pharmaceutical intermediate supplier, ensuring that final products meet stringent heavy metal specifications is a costly and time-consuming endeavor, often requiring multiple purification steps that erode overall yield and profitability.
The Novel Approach
In stark contrast to these conventional limitations, the methodology described in CN110467579B presents a paradigm shift by utilizing a metal-free, iodine-promoted cyclization strategy. This novel approach employs trifluoroethylimidoyl chloride and hydrazones as the primary building blocks, reacting them in the presence of sodium acetate and elemental iodine. The reaction proceeds smoothly in common organic solvents like 1,2-dichloroethane (DCE) at moderate temperatures around 80°C. Crucially, this process does not require rigorous anhydrous or anaerobic conditions, drastically reducing the operational complexity and infrastructure costs associated with the synthesis. The elimination of heavy metal catalysts not only enhances the environmental profile of the process but also simplifies the downstream workup, as there is no need for extensive scavenging or chromatographic removal of metal contaminants. This results in a cleaner crude product profile and higher isolated yields, making it an ideal candidate for cost reduction in API manufacturing.
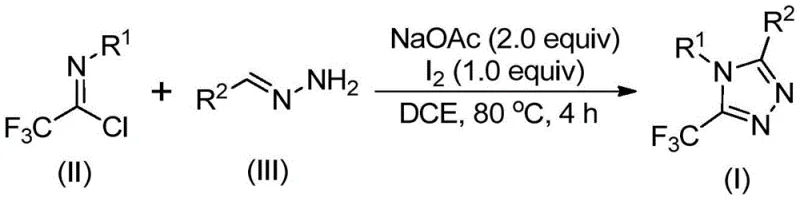
Mechanistic Insights into Iodine-Promoted Cyclization
The mechanistic pathway proposed for this transformation involves a sophisticated yet efficient sequence of bond-forming events driven by the dual role of the base and the iodine promoter. Initially, the reaction likely undergoes a base-promoted intermolecular carbon-nitrogen bond formation between the trifluoroethylimidoyl chloride and the hydrazone, generating a trifluoroacetamidine intermediate. This species then undergoes isomerization, setting the stage for the subsequent oxidative step. The addition of elemental iodine facilitates an oxidative iodination, creating a reactive iodine-bearing intermediate. This activation is critical, as it enables the subsequent intramolecular electrophilic substitution reaction. The nucleophilic attack by the nitrogen atom onto the activated carbon center leads to ring closure, followed by aromatization to yield the final stable 5-trifluoromethyl substituted 1,2,4-triazole core. This mechanism highlights the elegance of using simple halogens to drive complex heterocycle formation without the need for exotic ligands or high-energy inputs.
From an impurity control perspective, this mechanism offers distinct advantages. The stepwise nature of the reaction, coupled with the mild conditions, minimizes the formation of polymeric byproducts or decomposition species often seen in harsher radical trifluoromethylation processes. The use of sodium acetate as a mild base ensures that sensitive functional groups on the aromatic rings of the starting materials—such as esters, halides, or ethers—remain intact throughout the synthesis. This high functional group tolerance is essential for the commercial scale-up of complex pharmaceutical intermediates, where the preservation of stereochemistry and delicate moieties is non-negotiable. The robustness of this catalytic cycle ensures consistent batch-to-batch reproducibility, a key metric for supply chain reliability.
How to Synthesize 5-Trifluoromethyl-1,2,4-Triazoles Efficiently
The practical execution of this synthesis is designed for ease of operation, making it accessible for both laboratory discovery and pilot plant production. The protocol involves a straightforward one-pot procedure where the reagents are combined in a solvent system that effectively dissolves all components. The reaction timeline is optimized to balance conversion rates with energy consumption, typically requiring a total heating time of 3 to 6 hours depending on the specific substrate electronics. Detailed standardized synthesis steps, including precise stoichiometric ratios and workup procedures, are outlined in the technical guide below to ensure maximum yield and purity.
- Combine sodium acetate, trifluoroethylimidoyl chloride, and hydrazone in an organic solvent such as dichloroethane (DCE).
- Heat the reaction mixture to 80°C and maintain stirring for 2 to 4 hours to facilitate the initial condensation.
- Add elemental iodine to the system and continue heating for an additional 1 to 2 hours to complete the oxidative cyclization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route translates into tangible strategic benefits that extend beyond mere chemical curiosity. The primary value proposition lies in the drastic simplification of the supply chain for raw materials. Unlike specialized trifluoromethylating agents that may have limited suppliers and long lead times, the key starting materials for this process—hydrazones and trifluoroethylimidoyl chlorides—are derived from commodity chemicals like aromatic amines and aldehydes. This abundance ensures a stable supply base and mitigates the risk of production stoppages due to raw material shortages. Furthermore, the operational simplicity of the process reduces the burden on manufacturing facilities, allowing for faster turnaround times and more flexible production scheduling.
- Cost Reduction in Manufacturing: The economic impact of this technology is profound, primarily driven by the elimination of expensive transition metal catalysts and the associated purification costs. By removing the need for palladium or copper, manufacturers avoid the substantial expense of metal scavengers and the loss of product yield during metal removal steps. Additionally, the reaction operates at atmospheric pressure and moderate temperatures, which significantly lowers energy consumption compared to high-pressure hydrogenation or cryogenic reactions. The use of inexpensive elemental iodine as a promoter further drives down the cost of goods sold (COGS), enabling competitive pricing for high-purity pharmaceutical intermediates in a crowded market.
- Enhanced Supply Chain Reliability: The robustness of this synthetic method directly correlates with improved supply chain resilience. Because the reaction does not require stringent anhydrous or oxygen-free environments, it can be performed in standard glass-lined reactors without the need for specialized inert gas manifolds or drying trains. This flexibility allows for production in a wider range of facilities, diversifying the manufacturing network and reducing dependency on single-source high-tech sites. The high yields reported across a broad range of substrates indicate a reliable process that minimizes batch failures, ensuring consistent delivery schedules for downstream API manufacturers who depend on just-in-time inventory models.
- Scalability and Environmental Compliance: Scaling chemical processes from the bench to the plant often reveals hidden hazards, but this iodine-promoted cyclization appears inherently scalable. The exothermic profile is manageable, and the reagents are stable, reducing the risk of thermal runaways. From an environmental standpoint, the avoidance of heavy metals aligns perfectly with increasingly strict global regulations regarding waste disposal and residual limits in drug substances. The simplified waste stream, devoid of toxic metal sludge, lowers the cost of waste treatment and enhances the overall sustainability profile of the manufacturing process, a key factor for modern green chemistry initiatives.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity for technical teams evaluating this route for potential integration into their production pipelines.
Q: What are the key advantages of this iodine-promoted method over traditional trifluoromethylation?
A: This method avoids the use of expensive and toxic heavy metal catalysts and dangerous reagents like trifluorodiazoethane. It utilizes cheap, readily available starting materials (hydrazones and imidoyl chlorides) and operates under mild, non-anhydrous conditions, significantly simplifying purification and reducing safety risks.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the patent explicitly states that the method can be easily expanded to the gram level and beyond. The use of common solvents like DCE and standard heating conditions (80°C) makes it highly amenable to commercial scale-up without requiring specialized high-pressure or cryogenic equipment.
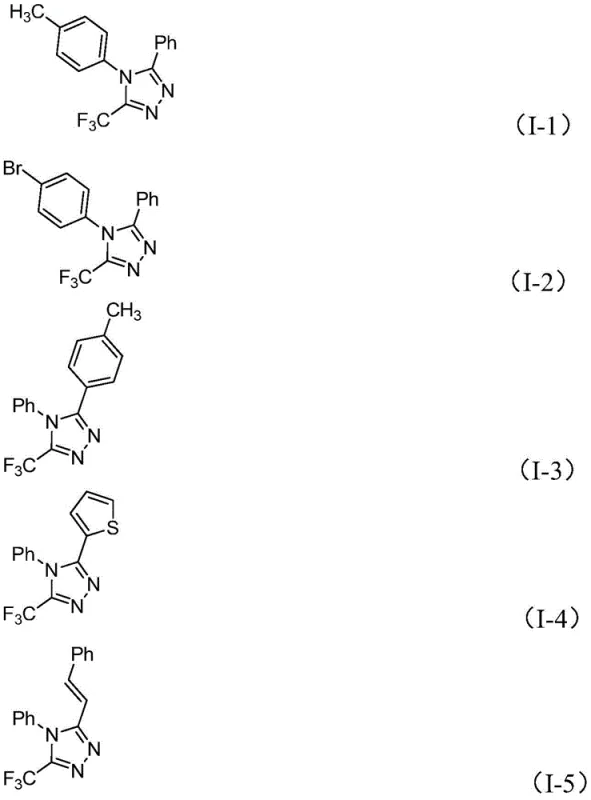
Q: What is the substrate scope for R1 and R2 groups in this synthesis?
A: The method demonstrates broad functional group tolerance. R1 can be various substituted aryl groups (e.g., methyl, methoxy, bromo, trifluoromethyl phenyl), while R2 accommodates alkenyl, aryl, and heteroaryl groups, allowing for the synthesis of diverse derivatives for drug discovery.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced heterocyclic intermediates play in the development of next-generation therapeutics. Our team of expert process chemists has thoroughly analyzed the technology disclosed in CN110467579B and is fully equipped to translate this academic innovation into commercial reality. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can move seamlessly from clinical trials to market launch. Our state-of-the-art facilities are designed to handle fluorine chemistry safely and efficiently, adhering to stringent purity specifications and operating within rigorous QC labs to guarantee the highest quality standards for every batch produced.
We invite you to collaborate with us to leverage this cost-effective and scalable synthesis for your specific drug candidates. By partnering with our technical procurement team, you can obtain a Customized Cost-Saving Analysis tailored to your volume requirements. We encourage you to reach out today to request specific COA data for our catalog compounds or to discuss route feasibility assessments for your proprietary molecules, ensuring a secure and efficient supply chain for your critical pharmaceutical intermediates.