Advanced Rhodium-Catalyzed Synthesis of Trifluoromethyl Enamines for Pharmaceutical Intermediates
Advanced Rhodium-Catalyzed Synthesis of Trifluoromethyl Enamines for Pharmaceutical Intermediates
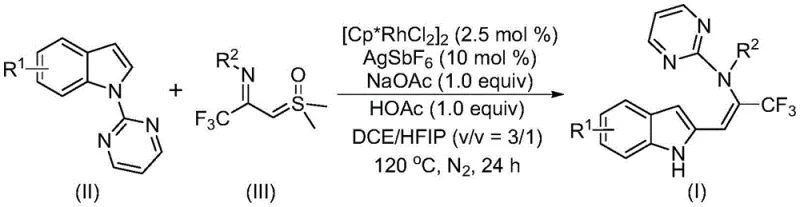
The landscape of modern pharmaceutical synthesis is constantly evolving, driven by the need for more efficient, cost-effective, and versatile methodologies to construct complex molecular architectures. A significant breakthrough in this domain is documented in Chinese Patent CN115925692A, which discloses a novel preparation method for trifluoromethyl-substituted enamine compounds. This technology represents a paradigm shift in how chemists approach the functionalization of indole scaffolds, utilizing a sophisticated rhodium-catalyzed carbon-hydrogen activation strategy coupled with a unique directing group migration mechanism. For R&D directors and procurement specialists alike, this patent offers a compelling solution for accessing high-value building blocks that are critical for the development of next-generation bioactive molecules and drug candidates. The ability to introduce both an indole skeleton and a trifluoromethyl group simultaneously through a tandem reaction sequence not only streamlines the synthetic pathway but also enhances the overall atom economy of the process.
Furthermore, the practical implications of this invention extend far beyond the laboratory bench, addressing critical pain points in the supply chain for fine chemical intermediates. By leveraging cheap and readily available starting materials such as indole derivatives and trifluoroacetimide sulfur ylides, the method drastically reduces the dependency on exotic or prohibitively expensive reagents that often bottleneck production schedules. The reaction operates under relatively standard thermal conditions, typically around 120°C, and utilizes a mixed solvent system that ensures high conversion rates and excellent yields, often exceeding 80% for a wide range of substrates. This level of efficiency and reliability makes it an attractive candidate for integration into existing manufacturing workflows, promising substantial cost reduction in API manufacturing while maintaining the rigorous purity standards required by the global pharmaceutical industry.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the introduction of enamine functional groups onto indole molecules has been a challenging endeavor, often relying on multi-step sequences that involve pre-functionalized substrates bearing specific directing groups. Traditional approaches frequently necessitate the use of ketene imines as substrates, undergoing metal-catalyzed directed carbon-hydrogen bond activation reactions that are limited by the structural diversity of the available starting materials. These conventional methods often suffer from restricted substrate scope, meaning that introducing different substituents on the aromatic rings can lead to significant drops in yield or complete reaction failure. Moreover, the requirement for specialized, pre-modified indole precursors adds layers of complexity and cost to the supply chain, as each variation requires a separate synthesis campaign before the actual enamine formation can even begin. This lack of flexibility severely limits the ability of medicinal chemists to rapidly explore structure-activity relationships (SAR) during the drug discovery phase.
The Novel Approach
In stark contrast, the methodology described in CN115925692A employs a transformative tandem reaction strategy that bypasses many of these historical limitations. By utilizing trifluoroacetimide sulfur ylides as both a trifluoromethyl source and an imine carbene precursor, the process enables a direct coupling with simple indole compounds. The core innovation lies in the rhodium-catalyzed C-H activation followed by a spontaneous 1,5-migration of the pyrimidine directing group. This elegant mechanism allows for the construction of highly functionalized enamine products in a single operational step, significantly reducing the number of unit operations required. The versatility of this approach is evidenced by its high tolerance for various functional groups, including halogens, alkyls, and electron-withdrawing groups, thereby enabling the synthesis of a diverse library of compounds from a common set of building blocks.
Mechanistic Insights into Rhodium-Catalyzed C-H Activation and Migration
To fully appreciate the technical sophistication of this process, one must delve into the proposed mechanistic pathway which underpins its high efficiency and selectivity. The reaction is initiated by the active cationic rhodium(III) species, generated in situ from the dimeric precursor [Cp*RhCl2]2 and the silver salt additive AgSbF6. This active catalyst coordinates with the nitrogen atom of the pyrimidine ring on the indole substrate, facilitating a directed ortho-metalation at the C2 position of the indole ring. This carbon-hydrogen activation step is crucial as it forms a stable rhodacycle intermediate, positioning the metal center perfectly for the subsequent insertion of the carbene species derived from the sulfur ylide. The precise control exerted by the catalyst ensures that the reaction occurs regioselectively, minimizing the formation of unwanted byproducts that would otherwise complicate downstream purification efforts.
Following the formation of the carbon-carbon bond, the intermediate undergoes a series of rearrangements that are key to the final product structure. Initially, an isomerization event generates an enamine motif, which is then followed by an intramolecular migration of the pyrimidine group. This 1,5-migration step is particularly noteworthy as it effectively transfers the directing group from the nitrogen atom to the carbon backbone, resulting in the final trifluoromethyl-substituted enamine product while regenerating the free indole NH proton. This mechanistic feature not only explains the high yields observed but also highlights the atom-economic nature of the transformation, as no atoms from the core scaffold are wasted. Understanding this mechanism is vital for process chemists aiming to optimize reaction parameters for commercial scale-up, as it identifies the catalyst loading and temperature as critical control points for maximizing throughput.
How to Synthesize Trifluoromethyl-Substituted Enamine Efficiently
Implementing this synthesis in a practical setting requires careful attention to the specific reaction conditions outlined in the patent data to ensure reproducibility and safety. The process involves combining the catalyst system, additives, and substrates in a specific molar ratio within a suitable organic solvent mixture. The use of a mixed solvent system comprising dichloroethane (DCE) and hexafluoroisopropanol (HFIP) is particularly important, as this combination has been shown to effectively promote the reaction progress and solubilize all reactants. Detailed standard operating procedures regarding the addition order, heating ramp rates, and workup protocols are essential for achieving the reported yields consistently.
- Combine the catalyst system comprising [Cp*RhCl2]2 and AgSbF6 with sodium acetate and acetic acid additives in a reaction vessel.
- Add the indole substrate and trifluoroacetimide sulfur ylide to a mixed solvent system of DCE and HFIP under nitrogen atmosphere.
- Heat the reaction mixture to 120°C for 24 hours, followed by filtration and column chromatography purification to isolate the target enamine.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthetic route offers tangible benefits that resonate deeply with procurement managers and supply chain heads who are tasked with optimizing costs and ensuring continuity of supply. The primary advantage lies in the accessibility of the raw materials; indole compounds and the requisite aromatic amines for preparing the sulfur ylides are commodity chemicals that are widely available from multiple global suppliers. This abundance mitigates the risk of supply chain disruptions that often plague processes reliant on bespoke or single-source reagents. Furthermore, the simplicity of the post-treatment process, which involves standard filtration and column chromatography, means that the technology can be integrated into existing purification infrastructure without the need for capital-intensive equipment upgrades.
- Cost Reduction in Manufacturing: The economic viability of this process is bolstered by the elimination of expensive pre-functionalized starting materials, which traditionally account for a significant portion of the COGS in complex molecule synthesis. By utilizing a direct C-H activation strategy, the number of synthetic steps is reduced, leading to lower labor costs, reduced solvent consumption, and decreased waste generation. Although specific percentage savings depend on the specific target molecule, the qualitative reduction in step count inherently drives down the overall manufacturing cost, making the final API more competitive in the marketplace.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions, which tolerate a wide array of functional groups, ensures that the supply of key intermediates remains stable even when sourcing variations occur. The ability to synthesize diverse analogs from a common platform means that inventory management can be streamlined, with bulk quantities of generic precursors stocked and converted to specific derivatives on demand. This flexibility is crucial for responding to fluctuating market demands in the pharmaceutical sector, where timelines for new drug approvals can be unpredictable.
- Scalability and Environmental Compliance: The patent explicitly notes that the reaction can be expanded to gram-level scales with high efficiency, suggesting a clear path toward kilogram and ton-scale production. The use of standard organic solvents and the absence of highly toxic heavy metals beyond the catalytic amount of rhodium simplify the environmental compliance landscape. Efficient catalyst turnover and the potential for catalyst recovery further align this process with green chemistry principles, reducing the environmental footprint associated with large-scale chemical manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this trifluoromethylation technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing a clear picture of what partners can expect when adopting this methodology for their own production needs.
Q: What are the key advantages of this Rhodium-catalyzed method over traditional enamine synthesis?
A: This method utilizes a directing group migration strategy that allows for the direct functionalization of inexpensive indole substrates without requiring pre-functionalized starting materials, significantly simplifying the synthetic route and reducing raw material costs.
Q: Is this process scalable for industrial production of pharmaceutical intermediates?
A: Yes, the patent explicitly demonstrates that the reaction conditions are robust and can be expanded to gram-level reactions with high efficiency, indicating strong potential for commercial scale-up in pharmaceutical manufacturing environments.
Q: What is the functional group tolerance of this trifluoromethylation reaction?
A: The methodology exhibits excellent functional group tolerance, accommodating various substituents such as halogens, alkyl groups, alkoxy groups, and nitro groups on both the indole and aryl rings, allowing for the synthesis of diverse bioactive molecular scaffolds.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl Enamine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the technologies described in CN115925692A and are uniquely positioned to bring these advanced synthetic routes to commercial reality. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial manufacturing is seamless and efficient. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of trifluoromethyl-substituted enamine meets the exacting standards required for pharmaceutical applications. Our commitment to quality and consistency makes us a trusted partner for companies seeking to secure their supply chains for critical drug intermediates.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis can be tailored to your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits specific to your target molecule. We encourage potential partners to contact us directly to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions that drive value and accelerate your drug development timelines. Let us collaborate to unlock the full potential of these powerful chemical building blocks.