Revolutionizing Opioid Alkaloid Production via Catalytic Asymmetric Total Synthesis
Revolutionizing Opioid Alkaloid Production via Catalytic Asymmetric Total Synthesis
The pharmaceutical industry has long relied on the extraction of morphine and codeine from opium poppies, a process fraught with agricultural volatility and regulatory complexity. However, a groundbreaking technical disclosure in patent CN109666030B introduces a paradigm shift towards efficient catalytic asymmetric total synthesis. This innovative methodology bypasses the limitations of natural extraction by constructing the complex morphinan skeleton from simple starting materials like 3-butyne-1-alcohol. The core breakthrough lies in the utilization of a specialized spirocyclic amine catalyst that drives a highly enantioselective intramolecular Michael addition. For R&D directors and procurement strategists, this represents a pivotal opportunity to secure a reliable API intermediate supplier capable of delivering high-purity opioids without the geopolitical risks associated with crop-based sourcing. The patent outlines a concise route that not only achieves superior stereocontrol but also opens avenues for synthesizing diversified morphine analogs essential for next-generation analgesic development.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the global supply of morphine and codeine has been inextricably linked to the cultivation of Papaver somniferum, creating a supply chain vulnerable to weather patterns, pestilence, and strict international narcotics controls. From a chemical perspective, extraction yields a fixed profile of alkaloids, severely restricting the ability of medicinal chemists to access novel structural variants for structure-activity relationship (SAR) studies. Furthermore, the isolation process often requires extensive purification to remove co-extracted impurities, leading to significant material loss and increased operational costs. Existing semi-synthetic methods often rely on chiral pool starting materials or resolution of racemates, which inherently cap the maximum theoretical yield at 50% or require expensive chiral resolving agents. These inefficiencies translate into higher manufacturing costs and longer lead times for high-purity pharmaceutical intermediates, posing a persistent challenge for supply chain heads aiming to optimize inventory and reduce dependency on single-source agricultural providers.
The Novel Approach
In stark contrast, the method disclosed in CN109666030B employs a convergent total synthesis strategy that constructs the chiral core de novo. By leveraging a spirocyclic amine organocatalyst, the process achieves exceptional enantioselectivity during the critical ring-forming step, effectively setting multiple stereocenters in a single operation. This approach eliminates the need for chiral resolution and allows for the precise installation of functional groups required for the final active pharmaceutical ingredients. The synthetic route is significantly shorter than many historical total syntheses, utilizing robust reactions such as Suzuki coupling, ozonolysis, and Birch reduction that are well-understood in process chemistry. This technological advancement offers a pathway for cost reduction in API manufacturing by streamlining the step count and improving overall atom economy. For procurement managers, this translates to a more predictable and scalable supply of codeine and morphine precursors, decoupling production from the uncertainties of agricultural harvests.
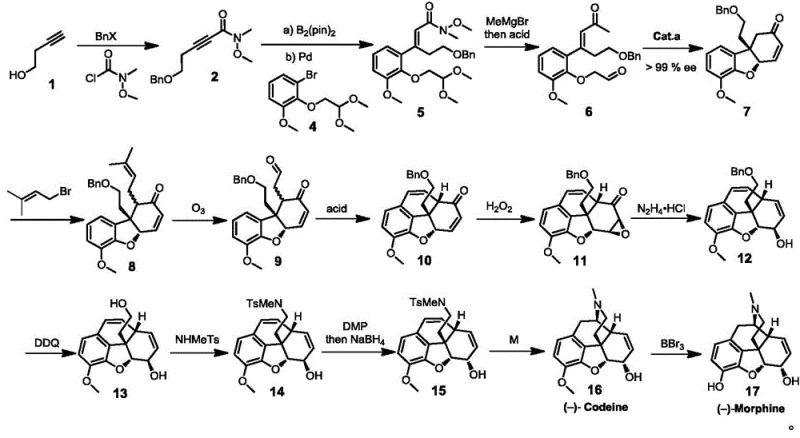
Mechanistic Insights into Spirocyclic Amine Catalyzed Cyclization
The heart of this synthetic innovation is the asymmetric organocatalytic cascade that transforms the linear precursor compound 6 into the tricyclic chiral compound 7. This transformation proceeds via an intramolecular Michael addition followed by acid-catalyzed dehydration cyclization. The spirocyclic amine catalyst, characterized by its rigid three-dimensional architecture, creates a highly defined chiral environment that directs the nucleophilic attack of the enamine intermediate onto the electron-deficient alkene. This precise spatial control ensures that the new carbon-carbon bonds are formed with >99% enantiomeric excess (ee), establishing the absolute configuration required for biological activity. The mechanism avoids the use of transition metals in this key step, which is a significant advantage for regulatory compliance regarding heavy metal residues in final drug substances. The subsequent acid-mediated cyclization locks the stereochemistry into the hydrogenated dibenzofuran scaffold, forming the core structure shared by both codeine and morphine.
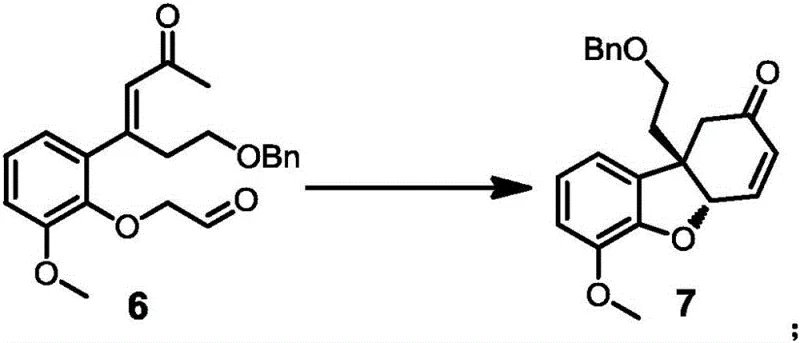
From an impurity control perspective, the high fidelity of the catalytic step drastically reduces the formation of diastereomeric byproducts that are notoriously difficult to separate in complex alkaloid synthesis. The use of mild reaction conditions, typically between -40°C and 30°C for the addition and 60°C to 120°C for cyclization, minimizes thermal degradation and side reactions such as polymerization or over-alkylation. Furthermore, the catalyst loading can be optimized between 5% and 30% molar weight, balancing reaction rate with cost efficiency. The robustness of this catalytic system allows it to tolerate various protecting groups and substituents, providing flexibility for the synthesis of analogs. For quality assurance teams, this mechanistic clarity means that critical process parameters (CPPs) can be tightly controlled to ensure consistent batch-to-batch quality, meeting the stringent purity specifications required for GMP manufacturing of controlled substances.
How to Synthesize Codeine Efficiently
The synthesis of codeine via this patented route involves a logical sequence of transformations designed to build complexity while maintaining stereochemical integrity. Starting from commercially available 3-butyne-1-alcohol, the process first installs necessary protecting groups and carbon frameworks through acylation and cross-coupling reactions. The pivotal step involves the aforementioned organocatalytic cyclization to establish the chiral center. Subsequent steps include allylation to extend the carbon chain, ozonolysis to generate aldehyde functionality, and an intramolecular Friedel-Crafts reaction to close the aromatic ring system. The detailed standardized synthetic steps see the guide below for specific reagent quantities and conditions optimized for laboratory and pilot scale operations.
- Synthesize compound 6 from 3-butyne-1-alcohol via protection, acylation, Suzuki coupling, and Grignard addition.
- Perform intramolecular Michael addition on compound 6 using a spirocyclic amine catalyst to form chiral compound 7 with high enantioselectivity.
- Convert compound 7 to codeine through allylation, ozonolysis, Friedel-Crafts reaction, epoxidation, Wharton rearrangement, and Birch reduction, followed by demethylation to morphine.
Commercial Advantages for Procurement and Supply Chain Teams
Adopting this catalytic asymmetric synthesis route offers profound strategic advantages for pharmaceutical supply chains, primarily by mitigating the risks associated with raw material sourcing. Traditional reliance on poppy straw concentrate subjects manufacturers to fluctuating market prices and regulatory quotas imposed by international narcotics control boards. By shifting to a synthetic origin, companies can establish a more resilient supply network that is immune to crop failures or geopolitical trade restrictions. This independence allows for better long-term capacity planning and inventory management, ensuring continuous availability of critical pain management medications even during global disruptions. Moreover, the ability to synthesize the core scaffold from petrochemical-derived starting materials provides a stable cost base that is less susceptible to the volatility of agricultural commodities.
- Cost Reduction in Manufacturing: The streamlined nature of this synthetic route directly contributes to significant cost optimization by reducing the total number of unit operations required to reach the final API. The elimination of chiral resolution steps, which typically discard half of the material, effectively doubles the theoretical yield of the chiral intermediate compared to racemic synthesis strategies. Additionally, the use of an organocatalyst rather than expensive transition metal complexes lowers the raw material cost per kilogram and simplifies the purification process by removing the need for rigorous metal scavenging. These efficiencies compound over large-scale production runs, resulting in substantial cost savings that can be passed down the supply chain or reinvested into further R&D initiatives.
- Enhanced Supply Chain Reliability: Synthetic manufacturing facilities can be located in diverse geographic regions, reducing the concentration risk inherent in crop-based sourcing which is limited to specific climatic zones. The starting materials, such as 3-butyne-1-alcohol and various benzyl halides, are commodity chemicals available from multiple global suppliers, ensuring redundancy in the upstream supply chain. This diversification enhances the reliability of supply for high-purity opioid intermediates, allowing procurement managers to negotiate better terms and secure long-term contracts with confidence. The predictability of chemical synthesis lead times also facilitates just-in-time manufacturing models, reducing the need for excessive safety stock and freeing up working capital.
- Scalability and Environmental Compliance: The reactions described in the patent, including Suzuki coupling and Birch reduction, are well-established in industrial organic chemistry and can be scaled from 100 kgs to 100 MT annual commercial production with known engineering controls. The process avoids the generation of large volumes of agricultural waste associated with poppy processing, aligning better with modern environmental sustainability goals. Furthermore, the high selectivity of the catalytic step reduces the solvent and energy consumption required for purification, lowering the overall environmental footprint of the manufacturing process. This compliance with green chemistry principles not only satisfies regulatory requirements but also enhances the corporate social responsibility profile of the manufacturing entity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthetic technology. They are derived from the specific experimental data and beneficial effects outlined in the patent documentation, providing clarity on feasibility and performance metrics. Understanding these details is crucial for technical teams evaluating the integration of this route into existing manufacturing portfolios.
Q: What is the primary advantage of this catalytic method over traditional extraction?
A: Unlike traditional extraction which relies on poppy cultivation and yields limited structural diversity, this catalytic asymmetric synthesis allows for the production of optically pure codeine and morphine independent of agricultural constraints, enabling the creation of diverse analog libraries for drug discovery.
Q: How does the spirocyclic amine catalyst impact product purity?
A: The spirocyclic amine catalyst facilitates a highly enantioselective intramolecular Michael addition, achieving greater than 99% ee in the formation of the critical chiral intermediate, thereby significantly reducing the burden of chiral separation downstream.
Q: Is this synthesis route scalable for industrial manufacturing?
A: Yes, the route utilizes robust reagents and standard organic transformations such as Suzuki coupling and Birch reduction. The catalytic step operates with low catalyst loading (5-30 mol%), making the process economically viable and suitable for commercial scale-up from kilograms to metric tons.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Codeine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this catalytic asymmetric synthesis technology for the global opioid market. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that complex synthetic routes like this are translated into robust industrial processes. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, including the precise measurement of enantiomeric excess and residual solvent levels. We are committed to delivering high-purity Codeine and Morphine intermediates that meet the exacting standards of international pharmacopoeias, supporting our partners in bringing life-saving analgesics to market faster and more efficiently.
We invite forward-thinking pharmaceutical companies to collaborate with us to leverage this advanced synthetic methodology. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions about securing a sustainable and cost-effective supply of these critical pharmaceutical ingredients.