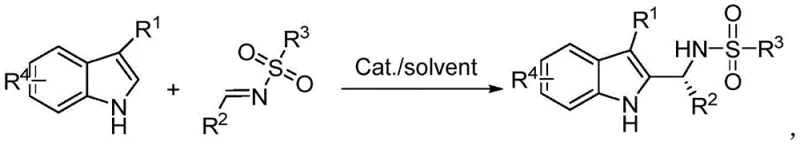
Scalable One-Step Synthesis of Chiral 2,3-Disubstituted Indoleamines for Global Pharmaceutical Supply Chains
The pharmaceutical and fine chemical industries are constantly seeking more efficient pathways to access complex chiral scaffolds, particularly those found in bioactive natural products and drug candidates. A significant breakthrough in this domain is documented in Chinese Patent CN113461589A, which discloses a robust preparation method for chiral 2,3-disubstituted indoleamine compounds. This technology represents a paradigm shift from traditional multi-step syntheses to a streamlined, one-step asymmetric organocatalytic process. By leveraging a novel chiral bissulfonimide catalyst, the method enables the direct coupling of 3-substituted indoles with aldimines via a Friedel-Crafts reaction. This innovation is critical for manufacturers aiming to enhance the purity and stereochemical integrity of their intermediate portfolios while simultaneously addressing the growing demand for sustainable and cost-effective chemical manufacturing processes.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of chiral 2,3-disubstituted indoleamine derivatives has been plagued by inefficiencies inherent in multi-step protocols. Prior art techniques typically relied on the asymmetric Friedel-Crafts reaction of 4,7-indolines with imines to generate 2-(4,7-indoline) amine derivatives, which subsequently required a separate oxidation step to yield the final 2-indoleamine structures. This two-step approach introduces significant operational complexity, including the need for additional reagents, extended reaction times, and intermediate isolation procedures that inevitably lead to material loss. Furthermore, the oxidation step often necessitates harsh conditions or specific oxidants that can compromise the stereochemical integrity of the chiral center, resulting in lower enantiomeric excess (ee) values. From a supply chain perspective, these additional unit operations increase the carbon footprint and elevate the cost of goods sold (COGS), making such routes less attractive for large-scale commercial production of high-value pharmaceutical intermediates.
The Novel Approach
In stark contrast, the methodology outlined in patent CN113461589A offers a direct, single-step solution that bypasses the need for pre-functionalized indoline substrates and subsequent oxidation. By employing a chiral bissulfonimide as an organic small-molecule catalyst, the reaction proceeds directly between readily available 3-substituted indoles and sulfonylimine compounds. This direct transformation not only drastically reduces the number of processing steps but also operates under remarkably mild conditions, typically around 35°C in common organic solvents like toluene. The elimination of the oxidation step removes a major bottleneck in the synthesis workflow, thereby enhancing the overall atom economy and process mass intensity (PMI). For procurement and supply chain managers, this translates to a more resilient manufacturing process with fewer points of failure, reduced solvent consumption, and a simplified purification workflow that facilitates faster time-to-market for critical drug intermediates.
Mechanistic Insights into Chiral Bissulfonimide Catalysis
The core of this technological advancement lies in the unique structural and electronic properties of the chiral bissulfonimide catalyst. Unlike traditional chiral phosphoric acids, the bissulfonimide catalyst described in the patent features a BINOL (1,1'-bi-2-naphthol) framework substituted with bulky sulfonimide groups, such as 3,5-bis(trifluoromethyl)phenyl groups. This specific architecture imparts a stricter C2 axis of symmetry and significantly stronger Brønsted acidity to the catalyst. Mechanistically, the catalyst functions by simultaneously activating both reaction partners through a dual hydrogen-bonding network. The acidic protons of the sulfonimide groups form hydrogen bonds with the nitrogen atom of the aldimine and the indole nitrogen, respectively. This precise organization creates a rigid chiral environment that effectively shields one face of the electrophile, forcing the nucleophilic attack of the indole to occur from a specific trajectory. This high degree of spatial control is what enables the reaction to achieve exceptional enantioselectivity, with experimental data showing ee values reaching up to 98% under optimized conditions.
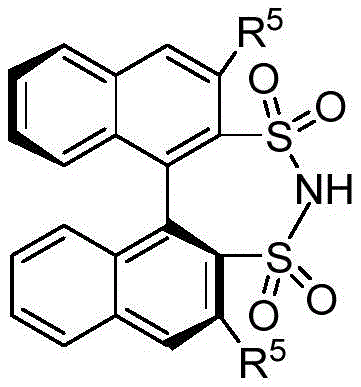
Furthermore, the mechanism ensures superior impurity control, which is a paramount concern for R&D directors overseeing quality assurance. The high specificity of the organocatalytic cycle minimizes the formation of regioisomers and racemic byproducts that are common in metal-catalyzed or non-selective acid-catalyzed reactions. Because the catalyst is metal-free, there is no risk of heavy metal contamination, a frequent issue with transition metal catalysts that requires expensive and time-consuming scavenging steps to meet regulatory limits (e.g., ICH Q3D guidelines). The reaction profile indicates that the catalyst loading can be kept relatively low (0.1 to 0.2 equivalents) while maintaining high turnover, suggesting a robust catalytic cycle that resists deactivation. This mechanistic robustness ensures consistent batch-to-batch reproducibility, a critical factor when scaling from laboratory grams to multi-kilogram commercial production runs.
How to Synthesize Chiral 2,3-Disubstituted Indoleamines Efficiently
The synthesis protocol described in the patent is designed for operational simplicity and scalability, making it highly attractive for process chemistry teams. The procedure involves preparing a mixed dispersion of the chiral catalyst in an organic solvent, followed by the sequential addition of the aldimine and indole substrates. The reaction is allowed to proceed under stirring at controlled temperatures, after which a simple aqueous quench and extraction workup yields the crude product. Purification is straightforward, typically requiring only silica gel column chromatography with standard eluent systems like ethyl acetate and petroleum ether. This streamlined workflow eliminates the need for specialized equipment or hazardous reagents, lowering the barrier to entry for manufacturing facilities. For a detailed breakdown of the standardized operating procedures and specific parameter ranges, please refer to the technical guide below.
- Prepare a mixed dispersion by stirring a chiral bissulfonimide catalyst (such as a BINOL-derived derivative) with an organic solvent like toluene.
- Sequentially add the aldimine substrate and the 3-substituted indole to the mixture and react under stirring at mild temperatures (e.g., 35°C).
- Quench the reaction with water, extract the crude product using ethyl acetate or dichloromethane, and purify via silica gel column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For stakeholders focused on the bottom line and supply continuity, the adoption of this organocatalytic route offers compelling economic and logistical benefits. The transition from a two-step metal-mediated or oxidation-dependent process to a direct one-step organocatalytic reaction fundamentally alters the cost structure of producing these valuable intermediates. By removing entire unit operations, manufacturers can realize substantial reductions in utility consumption, labor hours, and waste disposal costs. Moreover, the use of an organic small-molecule catalyst instead of precious metals mitigates the supply chain risks associated with fluctuating prices of rare earth elements or transition metals. This stability allows for more accurate long-term budgeting and pricing strategies for downstream pharmaceutical clients.
- Cost Reduction in Manufacturing: The most significant driver of cost savings is the elimination of the oxidation step and the associated reagents, which simplifies the bill of materials (BOM). Additionally, the absence of transition metals removes the necessity for expensive metal scavenger resins and the rigorous analytical testing required to certify low residual metal levels. This reduction in downstream processing complexity directly lowers the variable cost per kilogram of the final API intermediate. The mild reaction conditions (35°C) also imply lower energy demands for heating or cooling compared to cryogenic or high-temperature alternatives, further contributing to operational expenditure (OPEX) optimization without compromising yield or selectivity.
- Enhanced Supply Chain Reliability: The reagents utilized in this process, such as 3-substituted indoles and various aldimines, are commercially available and chemically stable, ensuring a reliable upstream supply. The catalyst itself, being an organic molecule, can be synthesized from abundant starting materials, reducing dependency on geopolitically sensitive mineral resources. This robustness in raw material sourcing translates to shorter lead times and higher confidence in meeting delivery schedules. For supply chain heads, the ability to source key intermediates from a process that is less susceptible to raw material volatility is a strategic advantage that enhances the overall resilience of the pharmaceutical supply network.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this method aligns perfectly with green chemistry principles. The use of toluene or other common solvents, combined with the lack of toxic heavy metals, simplifies waste stream management and reduces the environmental burden of the manufacturing site. The process generates less hazardous waste, lowering disposal costs and facilitating easier regulatory compliance. Furthermore, the simplicity of the reaction setup—requiring only standard stirring and temperature control—makes the technology highly scalable from pilot plant to full commercial production (100 MT scale) with minimal engineering modifications, ensuring a smooth technology transfer and rapid capacity ramp-up.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and claims within patent CN113461589A, providing a factual basis for evaluating the feasibility of this route for your specific project needs. Understanding these nuances is essential for making informed decisions about process adoption and supplier selection.
Q: What is the primary advantage of this synthesis method over conventional routes?
A: Unlike conventional methods that require a two-step process involving the oxidation of indoline derivatives, this patent describes a direct one-step asymmetric Friedel-Crafts reaction, significantly simplifying the workflow and improving overall efficiency.
Q: What type of catalyst is used to achieve high enantioselectivity?
A: The process utilizes a chiral bissulfonimide organocatalyst, specifically those with a BINOL framework, which offers stronger acidity and stricter C2 symmetry compared to traditional chiral phosphoric acids.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the reaction operates under mild conditions (e.g., 35°C) using common solvents like toluene and avoids toxic transition metals, making it highly adaptable for commercial scale-up and compliant with strict environmental standards.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral 2,3-Disubstituted Indoleamine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced organocatalytic technologies like the one described in CN113461589A for the production of high-value pharmaceutical intermediates. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this one-step synthesis are fully realized in a GMP-compliant manufacturing environment. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications and high enantiomeric excess, guaranteeing that every batch meets the exacting standards required by global regulatory bodies.
We invite you to collaborate with us to leverage this efficient synthesis route for your next-generation drug candidates. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how this technology can optimize your supply chain. Please contact our technical procurement team today to request specific COA data, route feasibility assessments, and a comprehensive quotation that reflects the true economic value of partnering with a supplier committed to innovation and quality excellence.