Revolutionizing Paclitaxel Production: A High-Yield Semi-Synthetic Route for Global Supply Chains
Revolutionizing Paclitaxel Production: A High-Yield Semi-Synthetic Route for Global Supply Chains
The global demand for Paclitaxel, a potent diterpenoid compound widely recognized for its exceptional anticancer properties and broad spectrum of activity, continues to outpace the limited supply derived from natural sources such as the bark of Taxus plants. Addressing this critical supply gap requires innovative semi-synthetic methodologies that offer superior efficiency and scalability compared to traditional extraction or total synthesis. The patent CN103450118A, published in December 2013, discloses a groundbreaking method for preparing Paclitaxel via semi-synthesis that fundamentally alters the conventional protection-deprotection sequence. By utilizing a cerium-based catalyst for selective acetylation, this process achieves high reaction yields under mild conditions, presenting a viable solution for the commercial scale-up of complex pharmaceutical intermediates. This technical insight report analyzes the mechanistic advantages and commercial implications of this novel route for industry stakeholders.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional semi-synthetic routes for Paclitaxel typically rely on 10-deacetylbaccatin III (10-DAB) as the starting material, necessitating a strict order of functional group manipulation to ensure regioselectivity. In standard protocols, the C-7 hydroxyl group, which is more reactive than the C-10 hydroxyl, must be protected first to prevent unwanted acetylation during subsequent steps. However, existing methods often suffer from significant drawbacks, including the use of harsh reagents for protection, low conversion yields around 85%, and the generation of substantial side products that complicate downstream purification. Furthermore, many prior art processes require rigorous inert gas protection and extended reaction times, creating bottlenecks that hinder cost reduction in API manufacturing. The reliance on column chromatography for purifying intermediates further exacerbates operational costs and environmental waste, making these conventional pathways less attractive for large-scale industrial adoption.
The Novel Approach
The methodology described in patent CN103450118A introduces a paradigm shift by reversing the traditional synthetic order, prioritizing the acetylation of the C-10 hydroxyl group before protecting the C-7 position. This is achieved through the strategic application of CeCl3.7H2O as a highly efficient and specific catalyst, which enables the selective acetylation of the C-10 position even in the presence of the more active C-7 hydroxyl. This innovation not only simplifies the synthetic workflow by avoiding the initial protection step but also ensures that the reaction proceeds rapidly at room temperature without the need for inert atmosphere protection. The result is a streamlined process characterized by fewer byproducts, simplified post-treatment procedures involving simple crystallization rather than chromatography, and overall milder reaction conditions that are ideally suited for reliable pharmaceutical intermediate supplier operations aiming for high throughput.
Mechanistic Insights into CeCl3-Catalyzed Selective Acetylation
The core chemical innovation of this process lies in the unique ability of the lanthanide salt CeCl3.7H2O to modulate the reactivity of hydroxyl groups on the baccatin skeleton. Under normal conditions, the nucleophilicity of the C-7 hydroxyl exceeds that of the C-10 hydroxyl, leading to non-selective acetylation if standard acylating agents are used alone. However, the presence of the cerium catalyst coordinates with the oxygen atoms in a manner that kinetically favors the acetylation of the C-10 position while leaving the C-7 hydroxyl untouched. This selectivity is crucial because it allows for the direct formation of Baccatin III from 10-DAB with reported molar yields reaching up to 94.4%, significantly higher than many traditional methods. The reaction is conducted in tetrahydrofuran (THF) at room temperature, demonstrating that high efficiency can be achieved without energy-intensive heating or cooling cycles, thereby enhancing the overall energy profile of the synthesis.
Following the selective acetylation, the process proceeds to protect the C-7 hydroxyl using chlorotriethylsilane (TES-Cl) in the presence of imidazole, yielding 7-TES-Baccatin III. This intermediate is then condensed with (4S,5R)-2,4-diphenyl-4,5-dihydrooxazole-5-carboxylic acid using DCC and DMAP coupling agents to form the Paclitaxel precursor. The final transformation involves a tandem deprotection and cyclization step where the oxazole ring is opened, and the TES group is simultaneously removed using dilute hydrochloric acid in a methanol/THF mixture. This concerted mechanism ensures that the final deprotection occurs smoothly at temperatures between 5°C and 25°C, minimizing thermal degradation of the sensitive taxane core and ensuring the integrity of the final high-purity Paclitaxel product.
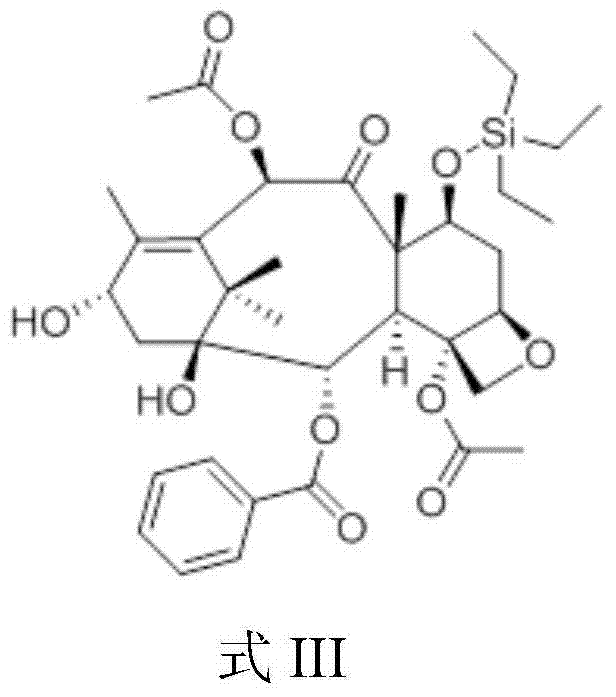
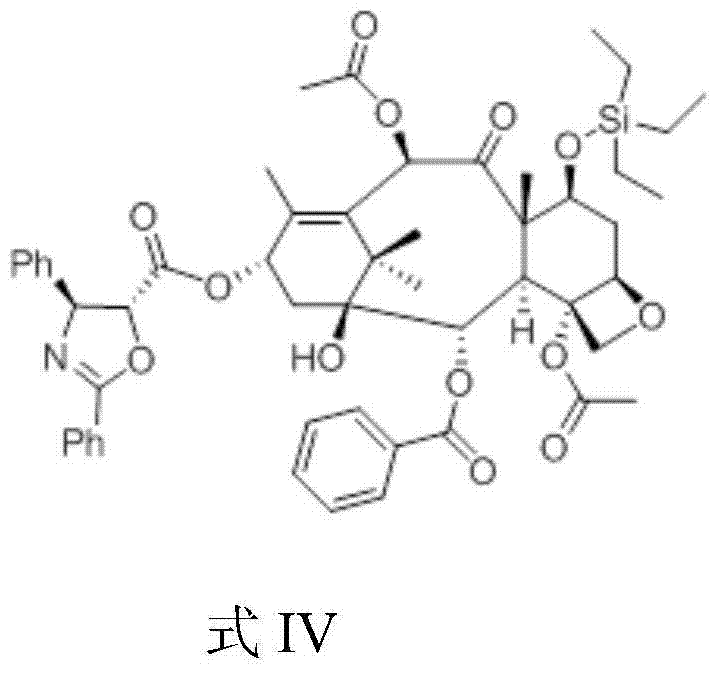
The structural integrity of the intermediates is paramount for the success of the final coupling reaction. As illustrated in the chemical structure of Formula III, the triethylsilyl group effectively shields the C-7 hydroxyl, preventing it from interfering with the esterification at the C-13 position. This protection strategy is robust yet labile enough to be removed under the mild acidic conditions of the final step. The subsequent condensation with the chiral oxazole acid creates the complex side chain characteristic of Paclitaxel, resulting in the precursor shown in Formula IV. The stereochemistry of the side chain is preserved throughout this process, which is critical for maintaining the biological activity of the final drug substance. The ability to construct this complex architecture with such high fidelity and yield underscores the robustness of this semi-synthetic pathway.
How to Synthesize Paclitaxel Efficiently
The synthesis of Paclitaxel via this patented route involves a logical sequence of four distinct chemical transformations that can be executed with standard laboratory equipment, facilitating easy technology transfer to pilot and production scales. The process begins with the dissolution of 10-DAB in THF, followed by the addition of the cerium catalyst and acetic anhydride to effect selective C-10 acetylation. After isolation of Baccatin III, the material is dissolved in DMF for the silylation step, followed by the coupling reaction in fresh DMF with the chiral acid. The final step utilizes a mixed solvent system of methanol and THF to effect global deprotection. For detailed operational parameters, stoichiometry, and workup procedures, please refer to the standardized synthesis guide below.
- Selective Acetylation: React 10-DAB with acetic anhydride in THF using CeCl3.7H2O catalyst at room temperature to form Baccatin III.
- C-7 Protection: Protect the 7-hydroxyl group of Baccatin III using chlorotriethylsilane and imidazole in DMF to obtain 7-TES-Baccatin III.
- Side-Chain Condensation: Condense 7-TES-Baccatin III with (4S,5R)-2,4-diphenyl-4,5-dihydrooxazole-5-carboxylic acid using DCC and DMAP to form the Paclitaxel precursor.
- Deprotection and Cyclization: Treat the precursor with dilute HCl in MeOH/THF to open the oxazole ring and remove the TES group, yielding final Paclitaxel.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of this semi-synthetic methodology offers transformative benefits that directly address the pain points of cost, lead time, and reliability in the pharmaceutical sector. By eliminating the need for column chromatography and relying instead on crystallization for purification, the process drastically reduces the consumption of silica gel and organic solvents, leading to substantial cost savings in raw materials and waste disposal. Furthermore, the ability to conduct reactions at room temperature without inert gas protection simplifies the engineering requirements for production reactors, allowing for faster batch turnover and reduced utility costs. These operational efficiencies translate into a more competitive pricing structure for the final API, enabling cost reduction in oncology drug manufacturing while maintaining stringent quality standards required by global regulatory bodies.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the avoidance of energy-intensive reaction conditions contribute to a leaner cost structure. Since the process avoids the use of rare gas protection and utilizes simple aqueous workups, the operational expenditure (OPEX) associated with each batch is significantly lowered. Additionally, the high molar yields reported in the patent examples, exceeding 90% in the final step, minimize the loss of valuable starting materials like 10-DAB, ensuring that the cost of goods sold (COGS) remains optimized for large-volume production runs.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions, which tolerate ambient temperatures and standard atmospheric pressure, reduces the risk of batch failures due to equipment malfunction or operator error. This reliability is crucial for maintaining a consistent supply of high-purity Paclitaxel to meet clinical and commercial demands. The use of readily available reagents such as CeCl3.7H2O and common solvents like THF and DMF ensures that the supply chain is not vulnerable to shortages of exotic or highly specialized chemicals, thereby securing the continuity of production schedules.
- Scalability and Environmental Compliance: The simplicity of the workup procedures, primarily involving filtration and crystallization, makes this process highly scalable from kilogram to multi-ton quantities without significant re-engineering. The reduction in solvent usage and the absence of heavy metal contaminants simplify the wastewater treatment process, aiding in compliance with increasingly strict environmental regulations. This eco-friendly profile not only mitigates regulatory risk but also aligns with the sustainability goals of modern pharmaceutical companies, making it an attractive option for green chemistry initiatives.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this semi-synthetic Paclitaxel process. These insights are derived directly from the experimental data and claims presented in the patent literature, providing a clear understanding of the process capabilities and limitations. Understanding these details is essential for R&D teams evaluating the feasibility of adopting this route for their own manufacturing pipelines.
Q: What is the primary advantage of using CeCl3.7H2O in this Paclitaxel synthesis?
A: The use of CeCl3.7H2O allows for highly selective acetylation of the C-10 hydroxyl group on 10-DAB without affecting the more reactive C-7 hydroxyl. This reverses the traditional protection order, eliminating the need for harsh protection steps first and significantly reducing byproduct formation.
Q: Does this method require column chromatography for purification?
A: No, one of the key commercial benefits of this patented process is that the intermediates and final product can be purified through simple crystallization and filtration. This eliminates the need for expensive and time-consuming column chromatography, drastically lowering production costs.
Q: What are the reaction conditions for the final deprotection step?
A: The final step involves reacting the Paclitaxel precursor in a mixed solvent of methanol and THF with 1:10 hydrochloric acid at mild temperatures between 5°C and 25°C. This simultaneously opens the oxazole ring and removes the C-7 protecting group.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Paclitaxel Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthetic routes in the production of life-saving oncology medications. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that complex molecules like Paclitaxel can be manufactured with consistency and precision. We are committed to delivering products that meet stringent purity specifications through our rigorous QC labs, which utilize state-of-the-art analytical instrumentation to verify the identity and quality of every batch. Our capability to implement advanced catalytic technologies, such as the cerium-mediated acetylation described in recent patents, positions us as a strategic partner for companies seeking to optimize their API supply chains.
We invite potential partners to engage with our technical procurement team to discuss how this innovative semi-synthetic route can be tailored to your specific volume requirements. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits of switching to this high-yield methodology. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions that will enhance the competitiveness and reliability of your pharmaceutical product portfolio.