Scalable Iron-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Global Pharmaceutical Manufacturing
Scalable Iron-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Global Pharmaceutical Manufacturing
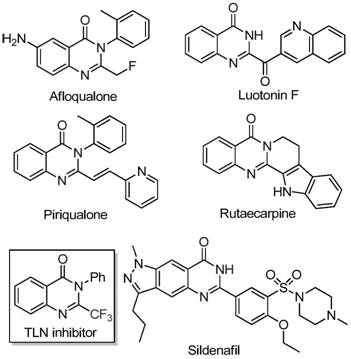
The pharmaceutical industry continuously seeks robust synthetic methodologies to access complex heterocyclic scaffolds that serve as the backbone for next-generation therapeutics. A significant breakthrough in this domain is detailed in patent CN111675662B, which discloses a highly efficient preparation method for 2-trifluoromethyl substituted quinazolinone compounds. This technology addresses critical bottlenecks in the synthesis of nitrogen-containing fused ring six-membered heterocycles, which are ubiquitous in molecular scaffolds exhibiting potent biological activities such as anti-cancer, anticonvulsant, and antifungal properties. By leveraging a novel iron-catalyzed cyclization strategy, this process transforms readily available starting materials into high-value intermediates with exceptional efficiency. For R&D directors and procurement specialists, understanding this pathway is essential for securing a reliable supply chain of advanced pharmaceutical intermediates while mitigating the costs associated with traditional precious metal catalysis.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinazolinone derivatives bearing trifluoromethyl functional groups has been fraught with significant technical and economic challenges. Literature reports indicate that conventional routes primarily rely on the cyclization of synthons containing trifluoromethyl groups, such as trifluoroacetic anhydride or ethyl trifluoroacetate, with substrates like anthranilamide or isatoic anhydride. While chemically feasible, these established methods are severely limited by harsh reaction conditions that often require stringent temperature controls and inert atmospheres. Furthermore, the substrates employed in these traditional pathways are frequently expensive and difficult to source in bulk quantities, leading to inflated production costs. The narrow substrate scope and generally low yields associated with these legacy processes create substantial barriers for commercial scale-up, making them less attractive for the manufacturing of cost-sensitive active pharmaceutical ingredients (APIs).
The Novel Approach
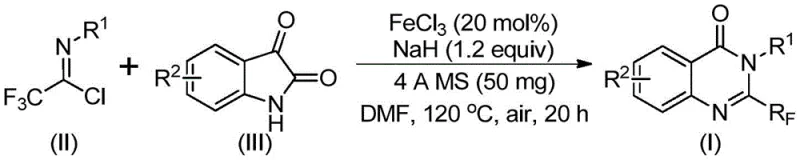
In stark contrast to these legacy techniques, the methodology outlined in patent CN111675662B introduces a paradigm shift by utilizing trifluoroethylimidoyl chloride and isatin as the primary building blocks. This innovative route employs a cheap metal iron catalyst, specifically ferric chloride, to drive the transformation under relatively mild conditions. The process demonstrates remarkable functional group tolerance, allowing for the incorporation of diverse substituents such as alkyl, halogen, and methoxy groups without compromising reaction efficiency. By operating in common organic solvents like DMF and utilizing air as the oxidant, this approach drastically simplifies the operational complexity. The ability to achieve high conversion rates with inexpensive reagents positions this technology as a superior alternative for the cost reduction in pharmaceutical intermediate manufacturing, directly addressing the pain points of supply chain volatility and raw material scarcity.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The core of this technological advancement lies in the intricate mechanistic pathway facilitated by the iron catalyst. During the reaction, an alkali-promoted carbon-nitrogen bond formation initially occurs between the trifluoroethylimidoyl chloride and the isatin derivative. This step generates a transient trifluoroacetamidine compound, which serves as a crucial intermediate in the catalytic cycle. Subsequently, the ferric chloride catalyst mediates a decarbonylation event, effectively removing the carbonyl oxygen from the isatin moiety. This is followed by a cyclization reaction that isomerizes the intermediate into the final stable 2-trifluoromethyl-substituted quinazolinone structure. The use of 4A molecular sieves plays a pivotal role in sequestering moisture, thereby protecting the sensitive imidoyl chloride species and ensuring the reaction proceeds with high fidelity. This mechanistic elegance allows for the construction of the quinazolinone core with precise regioselectivity.
From an impurity control perspective, this mechanism offers distinct advantages over radical-based or high-energy thermal processes. The stepwise nature of the bond formation and subsequent cyclization minimizes the generation of non-specific byproducts that are common in less controlled environments. The tolerance for various substituents on the aryl rings, including electron-withdrawing groups like nitro and halogens, suggests that the electronic properties of the substrates do not significantly hinder the catalytic turnover. This robustness implies a cleaner crude reaction profile, which simplifies downstream purification efforts. For quality assurance teams, this translates to a more predictable impurity spectrum, facilitating easier validation and regulatory compliance for the final drug substance. The ability to fine-tune the reaction by adjusting the molar ratios of ferric chloride and sodium hydride further empowers chemists to optimize the process for specific substrate classes.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
Implementing this synthesis route requires careful attention to reagent stoichiometry and thermal profiling to maximize yield and purity. The protocol involves a two-stage heating process where the initial mixing occurs at a lower temperature to facilitate the nucleophilic attack, followed by a higher temperature phase to drive the cyclization and decarbonylation to completion. The use of anhydrous conditions and specific solvent choices like DMF is critical for maintaining the integrity of the reactive intermediates. While the general procedure is straightforward, adherence to the specific molar ratios and reaction times detailed in the patent is essential for reproducing the high yields reported in the experimental examples. The detailed standardized synthesis steps for replicating this process in a pilot or production setting are provided in the guide below.
- Mix ferric chloride (20 mol%), sodium hydride (1.2 equiv), 4A molecular sieves, trifluoroethylimidoyl chloride, and isatin derivative in DMF solvent.
- Stir the reaction mixture at 40°C for approximately 10 hours to initiate the alkali-promoted bond formation.
- Heat the mixture to 120°C under air atmosphere for 18-20 hours to complete the iron-catalyzed decarbonylation and cyclization.
- Filter the reaction mixture, mix with silica gel, and purify via column chromatography to isolate the target quinazolinone compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this iron-catalyzed methodology represents a strategic opportunity to optimize the cost structure and reliability of the supply chain for quinazolinone-based APIs. The shift from expensive precious metal catalysts or specialized fluorinating agents to commodity chemicals like ferric chloride and isatin fundamentally alters the economic model of production. This transition not only lowers the direct material costs but also reduces the dependency on volatile markets for specialty reagents. Furthermore, the operational simplicity of the process, which does not require exotic equipment or extreme pressure conditions, enhances the overall agility of the manufacturing facility. These factors collectively contribute to a more resilient supply chain capable of meeting the rigorous demands of the global pharmaceutical market.
- Cost Reduction in Manufacturing: The replacement of costly catalysts and substrates with earth-abundant iron and bulk chemicals leads to a substantial decrease in the bill of materials. Eliminating the need for expensive trifluoroacetic anhydride removes a significant cost driver from the process. Additionally, the simplified workup procedure, which involves basic filtration and standard chromatography, reduces labor and solvent consumption compared to complex multi-step purifications. These cumulative efficiencies result in significant cost savings that can be passed down the value chain, enhancing the competitiveness of the final therapeutic product.
- Enhanced Supply Chain Reliability: The starting materials, specifically isatin derivatives and aromatic amines used to prepare the imidoyl chloride, are widely available from multiple global suppliers. This abundance mitigates the risk of single-source dependency and ensures continuity of supply even during market disruptions. The robustness of the reaction conditions means that production schedules are less likely to be impacted by minor variations in raw material quality or environmental factors. Consequently, manufacturers can maintain consistent inventory levels and meet delivery commitments with greater confidence, ensuring that downstream drug development programs remain on track.
- Scalability and Environmental Compliance: The protocol has been demonstrated to be effective at the gram level and is explicitly designed with industrial scale application in mind. The use of iron, a non-toxic and environmentally benign metal, aligns with green chemistry principles and simplifies waste management protocols. Unlike processes generating heavy metal waste streams that require costly disposal, the iron residues in this method are easier to handle and treat. This environmental compatibility facilitates smoother regulatory approvals and supports the sustainability goals of modern pharmaceutical enterprises, making it an ideal candidate for large-scale commercial production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis of 2-trifluoromethyl quinazolinones. These answers are derived directly from the technical specifications and beneficial effects described in the underlying patent documentation. They are intended to provide clarity on the feasibility, scope, and advantages of this specific catalytic system for stakeholders evaluating its potential integration into their manufacturing portfolios.
Q: What are the advantages of using iron catalysts over traditional methods for quinazolinone synthesis?
A: Traditional methods often rely on expensive trifluoroacetic anhydride or ethyl trifluoroacetate under severe conditions with low yields. The iron-catalyzed method described in patent CN111675662B utilizes cheap, earth-abundant FeCl3, offers mild conditions, high functional group tolerance, and significantly improved yields suitable for industrial scaling.
Q: Why is the introduction of a trifluoromethyl group important in drug design?
A: Incorporating a trifluoromethyl group into heterocyclic molecules like quinazolinones significantly enhances electronegativity, bioavailability, metabolic stability, and lipophilicity. These physicochemical improvements are critical for optimizing the pharmacokinetic profiles of potential anticancer, antifungal, and anti-inflammatory drug candidates.
Q: Is this synthesis method scalable for commercial production?
A: Yes, the patent explicitly states that the method can be expanded to the gram level and provides possibilities for industrial scale application. The use of readily available starting materials like isatin and simple post-treatment procedures involving filtration and chromatography supports robust commercial scalability.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
As the demand for fluorinated heterocycles continues to surge in the development of oncology and anti-infective therapies, partnering with an experienced CDMO is crucial for success. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions seamlessly from the laboratory to the marketplace. Our state-of-the-art facilities are equipped to handle the specific requirements of iron-catalyzed reactions, maintaining stringent purity specifications through our rigorous QC labs. We understand the critical nature of timeline and quality in drug development, and our team is dedicated to delivering high-purity intermediates that meet the exacting standards of the global pharmaceutical industry.
We invite you to leverage our technical expertise to optimize your supply chain for quinazolinone derivatives. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our implementation of this patented technology can enhance your project's viability. Let us collaborate to bring your next-generation therapeutics to patients faster and more efficiently.