Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial API Production
Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial API Production
The pharmaceutical and agrochemical industries are constantly seeking robust synthetic routes for heterocyclic scaffolds that offer enhanced metabolic stability and bioavailability. Patent CN112480015A discloses a groundbreaking method for synthesizing 2-trifluoromethyl substituted quinazolinones via a multicomponent one-pot strategy. This technology addresses critical bottlenecks in the production of high-value intermediates used in antifungal, antiviral, and anticancer drug development. The introduction of the trifluoromethyl group is particularly strategic, as it significantly modulates the electronegativity and lipophilicity of the parent molecule, leading to improved pharmacokinetic profiles. As illustrated in the structural diversity of known bioactive molecules, the quinazolinone core is a privileged structure found in numerous clinical candidates.
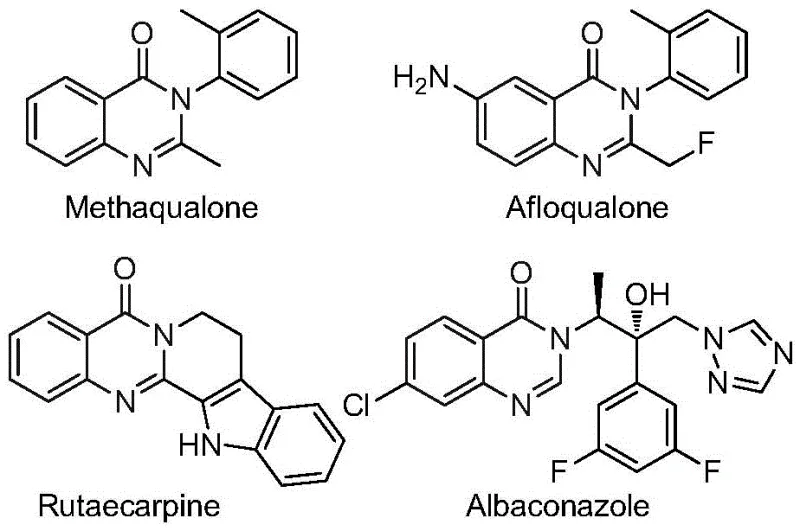
For R&D directors and process chemists, the ability to access these complex fused-ring nitrogen-containing heterocycles through a streamlined operation is a significant value proposition. The patent details a transition metal palladium-catalyzed carbonylation serial reaction that merges simplicity with high efficiency. By leveraging cheap and easily obtainable trifluoroethylimidoyl chloride and nitro compounds, this method widens the practicability of quinazolinone synthesis, making it an attractive option for reliable pharmaceutical intermediate suppliers aiming to optimize their catalog offerings.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the quinazolinone skeleton has been plagued by operational complexities and safety concerns. Traditional synthetic pathways often rely on the reduction and subsequent N-heterocyclic cyclization of nitro-substituted benzamides catalyzed by expensive ruthenium or platinum complexes. These processes frequently necessitate the use of high-pressure carbon monoxide gas, which introduces severe safety hazards and requires specialized high-pressure reactors that are not universally available in standard pilot plants. Furthermore, alternative methods involving iron-catalyzed condensations or palladium-catalyzed cyclizations of 2-bromoformanilides often suffer from narrow substrate scopes and low yields. The requirement for pre-activated substrates adds extra synthetic steps, increasing the overall cost of goods sold (COGS) and generating additional waste streams that complicate environmental compliance.
The Novel Approach
In stark contrast, the novel methodology presented in the patent utilizes a sophisticated yet operationally simple one-pot protocol. The core innovation lies in the use of nitro compounds as dual-purpose reagents: they act as the nitrogen source and undergo in-situ reduction to amines, which then participate in the cyclization. This eliminates the need for isolating sensitive amine intermediates. The reaction employs a palladium chloride catalyst coordinated with a dppp ligand and utilizes molybdenum hexacarbonyl as a safe, solid carbon monoxide surrogate. This approach allows the reaction to proceed at atmospheric pressure conditions relative to gas handling, significantly lowering the barrier to entry for scale-up. The general reaction scheme highlights the convergence of trifluoroethylimidoyl chloride and various nitroarenes to form the target scaffold efficiently.
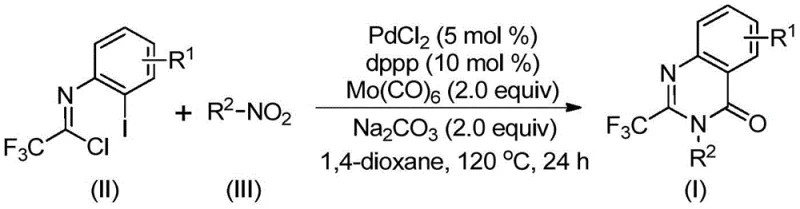
This novel route not only simplifies the workflow but also enhances the atom economy of the process. By avoiding high-pressure gas cylinders and expensive noble metals like ruthenium, the method offers a pathway for substantial cost reduction in pharmaceutical intermediate manufacturing. The compatibility with a wide range of functional groups means that a single optimized protocol can be applied to generate a library of derivatives, accelerating the lead optimization phase for drug discovery teams.
Mechanistic Insights into Pd-Catalyzed Carbonylative Cyclization
Understanding the mechanistic underpinnings of this transformation is crucial for process optimization and impurity control. The reaction initiates with the reduction of the nitro compound to the corresponding amine by molybdenum hexacarbonyl under heating conditions. This generated amine then undergoes an alkali-promoted coupling with the trifluoroethylimidoyl chloride to form a trifluoroacetamidine derivative intermediate. Subsequently, the palladium catalyst inserts into the carbon-iodine bond of the imidoyl chloride moiety, forming a divalent palladium species. The molybdenum hexacarbonyl serves as the CO source, releasing carbon monoxide which inserts into the carbon-palladium bond to generate an acyl palladium intermediate.
The final stages involve the formation of a palladium-nitrogen bond promoted by the base, leading to a seven-membered ring palladium intermediate. Reductive elimination from this species yields the final 2-trifluoromethyl-substituted quinazolinone compound and regenerates the active catalyst. This intricate cascade ensures high chemoselectivity, minimizing the formation of side products such as ureas or unreacted amidines. The ability to tolerate diverse substituents, as shown in the specific examples where R1 can be H, methyl, F, Cl, Br, or CF3, and R2 can be aryl, alkyl, or cycloalkyl, underscores the robustness of the catalytic cycle. This mechanistic clarity allows for precise tuning of reaction parameters to maximize purity, a key concern for high-purity OLED material or API intermediate production.
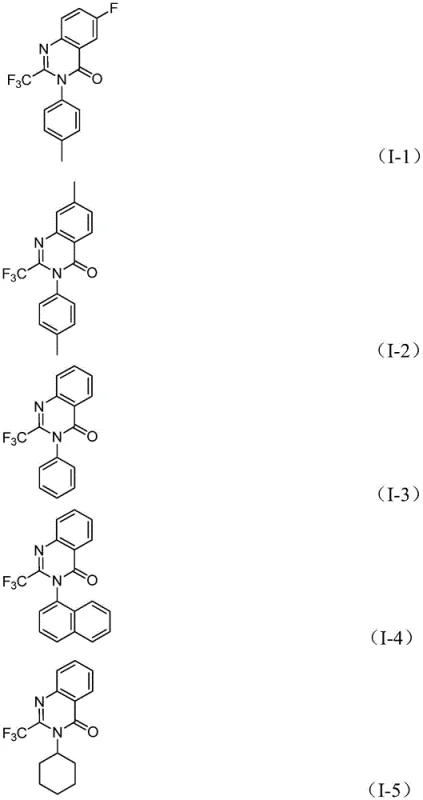
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
The execution of this synthesis requires careful attention to reagent stoichiometry and temperature control to ensure maximum conversion. The patent outlines a standardized procedure where palladium chloride, the dppp ligand, sodium carbonate, and Mo(CO)6 are combined with the substrates in an aprotic solvent like dioxane. The reaction is typically heated to 120°C for a duration of 16 to 30 hours. Post-reaction processing involves simple filtration followed by silica gel mixing and column chromatography purification. For detailed operational parameters and specific molar ratios optimized for different substrates, please refer to the standardized synthesis guide below.
- Mix palladium chloride, dppp ligand, sodium carbonate, Mo(CO)6, trifluoroethylimidoyl chloride, and nitro compound in an organic solvent like dioxane.
- Heat the reaction mixture to 120°C and stir for 16 to 30 hours to allow the carbonylation and cyclization sequence to complete.
- Filter the reaction mixture, mix with silica gel, and purify via column chromatography to isolate the target quinazolinone compound.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this technology offers compelling advantages that directly impact the bottom line and operational resilience. The shift from high-pressure gas reactions to solid-state CO surrogates drastically simplifies the infrastructure requirements for manufacturing facilities. This reduction in complexity translates to lower capital expenditure (CAPEX) for new production lines and reduced maintenance costs for existing ones. Furthermore, the reliance on commercially available and inexpensive starting materials like nitro compounds and trifluoroethylimidoyl chloride ensures a stable and continuous supply chain, mitigating the risks associated with sourcing exotic or custom-synthesized precursors.
- Cost Reduction in Manufacturing: The elimination of expensive ruthenium or platinum catalysts in favor of a more economical palladium system significantly lowers raw material costs. Additionally, the one-pot nature of the reaction removes the need for intermediate isolation and purification steps, which are often the most labor-intensive and solvent-consuming parts of a synthesis. This streamlining leads to substantial cost savings in terms of solvent usage, waste disposal, and man-hours, making the process highly competitive for large-scale production.
- Enhanced Supply Chain Reliability: The substrates utilized in this method, particularly the nitro compounds and the imidoyl chlorides, are commodity chemicals with established global supply chains. This availability reduces the lead time for high-purity pharmaceutical intermediates, allowing manufacturers to respond quickly to market demands. The robustness of the reaction conditions also means that batch-to-batch variability is minimized, ensuring consistent quality and reliable delivery schedules for downstream customers.
- Scalability and Environmental Compliance: The use of molybdenum hexacarbonyl as a CO source avoids the regulatory hurdles and safety protocols associated with storing and transporting high-pressure carbon monoxide gas. This makes the process inherently safer and easier to scale from gram-level laboratory synthesis to multi-ton commercial production. The simplified workup procedure, involving filtration and chromatography, generates less hazardous waste compared to traditional methods, aligning with modern green chemistry principles and facilitating easier environmental compliance.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation of this synthesis method. These answers are derived directly from the experimental data and specifications provided in the patent documentation, ensuring accuracy and relevance for technical decision-makers evaluating this technology for adoption.
Q: What are the advantages of using nitro compounds over amines in this synthesis?
A: Nitro compounds serve as inexpensive and readily available starting materials that undergo in-situ reduction, eliminating the need for separate reduction steps and harsh pre-activation conditions often required for amine substrates.
Q: Can this method tolerate diverse functional groups on the aromatic ring?
A: Yes, the protocol demonstrates excellent substrate compatibility, successfully accommodating halogens (F, Cl, Br), alkyl groups, and trifluoromethyl substituents at ortho, meta, and para positions without significant yield loss.
Q: Is high-pressure carbon monoxide gas required for this reaction?
A: No, the method utilizes molybdenum hexacarbonyl (Mo(CO)6) as a solid carbon monoxide substitute, which releases CO under heating conditions, thereby avoiding the safety hazards and specialized equipment associated with high-pressure CO gas.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient heterocycle synthesis in the development of next-generation therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries can be seamlessly transitioned into industrial reality. We are committed to delivering products that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify every batch.
We invite potential partners to engage with our technical procurement team to discuss how this advanced synthesis route can be integrated into your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this methodology. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your specific project requirements, ensuring a partnership built on transparency and technical excellence.