Advanced Asymmetric Synthesis of Trifluoromethyl-Containing Polyfunctional Cyclopentenone Derivatives for Commercial Scale-Up
Introduction to Patent CN113501764B Technology
The pharmaceutical industry continuously seeks robust methodologies for constructing chiral scaffolds that possess metabolic stability and biological activity. Patent CN113501764B introduces a groundbreaking asymmetric synthesis method for preparing trifluoromethyl-containing polyfunctional cyclopentenone derivatives. These compounds serve as critical building blocks in modern drug discovery, particularly due to the unique properties imparted by the trifluoromethyl group, such as enhanced lipophilicity and metabolic resistance. The core innovation lies in a trifluoromethyl radical-mediated three-component aza-Piancatelli rearrangement reaction. By utilizing a sophisticated combination of monovalent copper, a chiral Bronsted acid, and an achiral Lewis acid, this technology enables the direct assembly of complex chiral architectures from readily available furan olefins, Togni reagents, and arylamine compounds. This represents a significant leap forward in synthetic efficiency, providing a reliable pathway for generating high-value intermediates that were previously difficult to access with high stereocontrol.
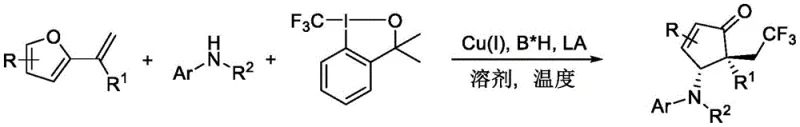
The strategic importance of this patent cannot be overstated for organizations focused on developing next-generation therapeutics. Traditional methods often struggle with the simultaneous introduction of the trifluoromethyl moiety and the establishment of multiple stereocenters in a single operation. This novel catalytic system overcomes these hurdles by orchestrating a cascade reaction that builds molecular complexity rapidly. The reaction conditions are notably mild, typically proceeding at moderate temperatures in common organic solvents like chloroform. This accessibility makes the technology highly attractive for both early-stage medicinal chemistry campaigns and later-stage process development. For a reliable pharmaceutical intermediate supplier, mastering such transformations is essential to meeting the evolving demands of clients who require diverse, high-purity building blocks for their pipeline programs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of chiral cyclopentenone skeletons has relied heavily on furyloxonium ion-directed asymmetric rearrangement reactions. However, these conventional pathways have been predominantly limited to two-component rearrangements based on furfuryl alcohol substrates. This restriction severely curtails the functional group compatibility of the reaction, as furfuryl alcohols can be sensitive to various reaction conditions and may not tolerate the diverse substituents required for specific drug targets. Furthermore, the existing literature lacked efficient methods for introducing a trifluoromethyl group directly into the side chain of the cyclopentenone ring during the rearrangement process. Chemists were often forced to employ multi-step sequences involving protection and deprotection strategies, or post-functionalization modifications that could erode overall yield and stereochemical integrity. These inefficiencies translate into higher costs and longer lead times for producing high-purity intermediates, creating bottlenecks in the supply chain for active pharmaceutical ingredients.
The Novel Approach
In stark contrast, the methodology described in CN113501764B utilizes furan olefins instead of furfuryl alcohols, engaging them in a three-component coupling with Togni reagents and arylamines. This shift in substrate strategy dramatically expands the chemical space accessible to synthetic chemists. The use of Togni reagents as the trifluoromethyl source allows for the direct installation of the CF3 group with high regioselectivity. The synergistic catalytic system, comprising Cu(I), a chiral phosphoric acid derivative, and a lanthanide Lewis acid, ensures exceptional control over both enantioselectivity and diastereoselectivity. This approach eliminates the need for pre-functionalized chiral starting materials, thereby streamlining the synthesis. For procurement managers, this means cost reduction in API manufacturing is achievable through shorter synthetic routes and reduced waste generation. The ability to tolerate a wide range of electron-withdrawing and electron-donating groups on the arylamine component further underscores the versatility of this novel approach for generating diverse libraries of compounds.
Mechanistic Insights into Cu-Catalyzed Asymmetric Aza-Piancatelli Rearrangement
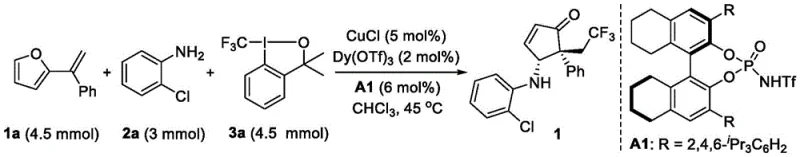
The success of this transformation hinges on the precise interplay between the radical generator and the chiral environment created by the catalysts. The mechanism initiates with the monovalent copper species activating the Togni reagent to generate a trifluoromethyl radical. This radical adds to the furan olefin substrate, triggering a cascade of electronic rearrangements. The chiral Bronsted acid, specifically the BINOL-derived phosphoric acid A1 depicted in the patent, plays a pivotal role in organizing the transition state. Through hydrogen bonding interactions, it activates the imine intermediate formed from the arylamine, ensuring that the subsequent cyclization occurs with high facial selectivity. Simultaneously, the achiral Lewis acid, such as dysprosium triflate, coordinates with oxygen atoms to stabilize reactive intermediates and accelerate the rearrangement step. This dual-activation mode is critical for achieving the reported enantiomeric excess values of up to 95 percent.
From an impurity control perspective, this mechanistic pathway offers distinct advantages. The high diastereoselectivity observed, often exceeding 20:1 dr, implies that the formation of unwanted stereoisomers is effectively suppressed at the molecular level. This reduces the burden on downstream purification processes, which is a key consideration for R&D directors focused on process robustness. The radical nature of the initial step might raise concerns about side reactions, but the optimized reaction conditions and the specific choice of ligands minimize radical dimerization or non-productive pathways. The result is a clean reaction profile that yields the desired chiral multifunctional cyclopentenone as the major product. Understanding these mechanistic nuances allows process chemists to fine-tune reaction parameters, such as temperature and stoichiometry, to maximize efficiency while maintaining the stringent purity specifications required for pharmaceutical applications.
How to Synthesize Trifluoromethyl Cyclopentenone Efficiently
Implementing this synthesis route requires careful attention to the preparation of the catalytic system and the order of reagent addition to ensure optimal performance. The patent outlines a standardized protocol that begins with the drying of the reaction vessel and the establishment of an inert argon atmosphere, which is crucial for preventing the oxidation of the copper catalyst. The sequential addition of the Togni reagent, arylamine, and furan olefin at low temperature helps to manage the exothermicity of the radical initiation step before warming the mixture to the reaction temperature. Detailed standardized synthesis steps are provided in the guide below to assist technical teams in replicating these results accurately.
- Prepare the catalytic system by combining CuCl, Dy(OTf)3, and chiral Bronsted acid A1 in a reaction vessel under inert atmosphere.
- Add the solvent chloroform and cool the mixture to 0°C before sequentially introducing Togni reagent, arylamine, and furan olefin substrates.
- Warm the reaction to 45°C and stir for 8-36 hours, monitoring progress via TLC, followed by purification using silica gel column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For supply chain leaders and procurement specialists, the adoption of this technology presents compelling economic and operational benefits. The primary advantage lies in the simplification of the synthetic route. By consolidating multiple bond-forming events into a single pot, the process significantly reduces the number of unit operations required. This consolidation leads to substantial cost savings associated with solvent usage, energy consumption, and labor. Moreover, the starting materials, including furan olefins and arylamines, are generally commercially available or easily synthesized from commodity chemicals, ensuring a stable and reliable supply chain. The elimination of precious metal catalysts in favor of more abundant copper salts further contributes to cost reduction in manufacturing, making the process economically viable for large-scale production without compromising on quality.
- Cost Reduction in Manufacturing: The streamlined nature of this three-component reaction directly impacts the cost of goods sold. By avoiding multi-step sequences and protecting group manipulations, the overall material throughput is improved. The high yields reported in the patent examples mean that less raw material is wasted, enhancing the atom economy of the process. Additionally, the use of standard purification techniques like silica gel chromatography, which is well-understood and scalable, avoids the need for specialized and expensive separation technologies. These factors combine to create a highly efficient manufacturing process that lowers the barrier to entry for producing complex fluorinated intermediates, allowing companies to allocate resources more effectively across their portfolio.
- Enhanced Supply Chain Reliability: Dependence on exotic or hard-to-source reagents can introduce significant risk into the supply chain. This methodology relies on robust and widely available reagents such as Togni reagents and simple aniline derivatives. The resilience of the catalytic system to varying substrate electronics means that a single set of conditions can often be applied to a broad range of analogues. This flexibility allows suppliers to respond quickly to changes in demand or specific structural requirements from clients. Reducing lead time for high-purity intermediates becomes feasible because the synthesis is not bottlenecked by the availability of niche starting materials. The demonstrated stability of the reaction conditions also implies consistent batch-to-batch quality, which is paramount for maintaining long-term supply agreements with pharmaceutical partners.
- Scalability and Environmental Compliance: Scaling chemical processes from the bench to the plant floor often reveals hidden challenges, but the patent data includes successful scale-up experiments that validate the commercial potential of this route. The reaction proceeds efficiently at larger scales with maintained stereoselectivity, indicating that heat and mass transfer issues are manageable. From an environmental standpoint, the reduction in step count inherently reduces the volume of chemical waste generated. The use of chloroform as a solvent is noted, but opportunities exist to substitute this with greener alternatives in future process optimizations without altering the core chemistry. The ability to produce complex chiral molecules with high efficiency aligns with modern green chemistry principles, helping companies meet increasingly stringent environmental regulations while maintaining production capacity.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this asymmetric synthesis technology. These answers are derived directly from the experimental data and claims within the patent documentation, providing clarity for technical teams evaluating this route for their specific projects. Understanding the boundaries and capabilities of the reaction helps in making informed decisions about process adoption and resource allocation.
Q: What is the enantioselectivity achieved in this synthesis method?
A: The patented method achieves excellent enantioselectivity, with enantiomeric excess (ee) values reaching up to 95% for various trifluoromethyl-containing cyclopentenone derivatives.
Q: Can this process be scaled up for industrial production?
A: Yes, the patent demonstrates successful scale-up experiments, maintaining high yield and stereoselectivity when increasing reactant quantities significantly beyond milligram scales.
Q: What are the key advantages over conventional furfuryl alcohol methods?
A: This three-component approach using furan olefins offers broader functional group compatibility and avoids the limitations of traditional two-component rearrangements restricted to furfuryl alcohol substrates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl Cyclopentenone Supplier
The technological advancements detailed in CN113501764B represent a significant opportunity for the pharmaceutical industry to access high-quality chiral intermediates more efficiently. NINGBO INNO PHARMCHEM stands at the forefront of translating such innovative academic and patent research into practical, commercial realities. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facilities are equipped with rigorous QC labs capable of verifying the stringent purity specifications and stereochemical integrity required for advanced drug candidates. We understand that the transition from milligram-scale discovery to ton-scale manufacturing requires not just chemical expertise but also deep engineering knowledge to ensure safety and consistency.
We invite you to collaborate with us to leverage this cutting-edge synthesis method for your pipeline. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline. By partnering with us, you gain access to specific COA data and route feasibility assessments that can de-risk your supply chain. Whether you need rapid gram-scale delivery for preclinical studies or multi-ton support for clinical trials, our commitment to quality and reliability ensures that your project moves forward without interruption. Contact us today to discuss how we can support your needs for high-purity pharmaceutical intermediates and drive your development programs forward.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →