Scalable Production of 2-Trifluoromethyl Quinazolinones via Iron Catalysis for Global Pharma
Scalable Production of 2-Trifluoromethyl Quinazolinones via Iron Catalysis for Global Pharma
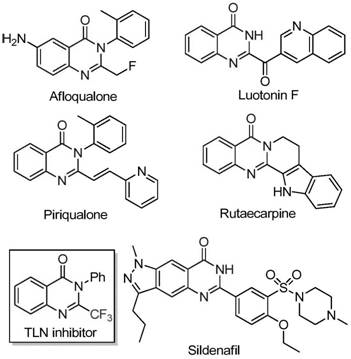
The pharmaceutical industry continuously seeks robust synthetic methodologies for constructing nitrogen-containing heterocycles, particularly quinazolinones, due to their pervasive presence in bioactive molecules. As detailed in the recent patent CN111675662B, a novel preparation method for 2-trifluoromethyl substituted quinazolinone compounds has been disclosed, offering a significant advancement over traditional synthetic routes. This technology leverages the unique electronic properties of the trifluoromethyl group to enhance the lipophilicity and metabolic stability of drug candidates, addressing a critical need in modern medicinal chemistry. The introduction of fluorine atoms into heterocyclic scaffolds is a well-established strategy to improve binding affinity and pharmacokinetic profiles, making these intermediates highly valuable for the development of anticancer, anticonvulsant, and anti-inflammatory agents. By utilizing readily available starting materials and an inexpensive iron catalyst, this invention provides a sustainable and economically attractive pathway for the commercial production of these high-value pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinazolinones bearing trifluoromethyl functionalities has relied heavily on the cyclization of synthons such as anthranilamide, anthranilic acid, or isatoic anhydride with trifluoroacetic anhydride or ethyl trifluoroacetate. While these methods are chemically feasible, they suffer from significant drawbacks that hinder their application in large-scale manufacturing. The reaction conditions are often severe, requiring high temperatures or strong bases that can degrade sensitive functional groups, leading to complex impurity profiles that are difficult to separate. Furthermore, the substrates used in these traditional protocols, particularly specialized trifluoromethyl synthons, can be prohibitively expensive and difficult to source in bulk quantities, creating bottlenecks in the supply chain. The narrow substrate scope of many prior art methods also limits the ability of medicinal chemists to rapidly generate diverse libraries of analogues for structure-activity relationship (SAR) studies, slowing down the drug discovery process.
The Novel Approach
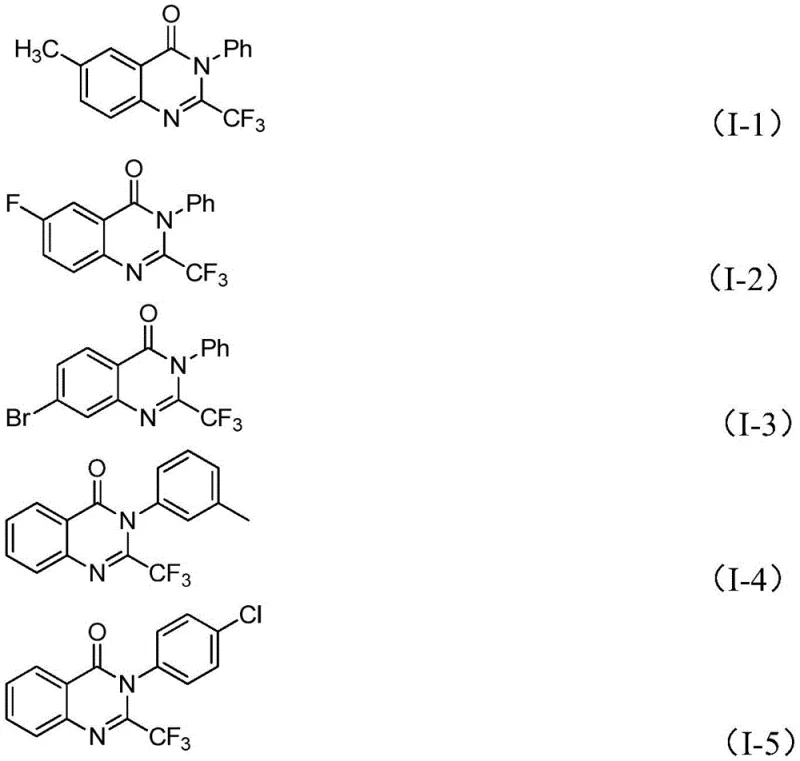
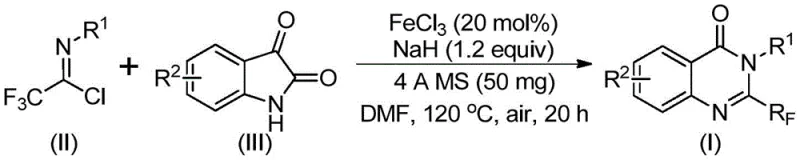
In stark contrast to these legacy methods, the disclosed invention utilizes a streamlined approach involving the reaction of trifluoroethylimidoyl chloride with isatin derivatives. This strategy capitalizes on the high reactivity of the imidoyl chloride species and the accessibility of isatins to construct the quinazolinone core efficiently. The process is catalyzed by ferric chloride, an abundant and low-cost transition metal salt, which replaces expensive noble metals often found in cross-coupling reactions. This shift not only drastically reduces the bill of materials but also aligns with green chemistry principles by minimizing the environmental footprint associated with precious metal mining and processing. The method exhibits remarkable functional group tolerance, successfully accommodating electron-donating and electron-withdrawing groups such as methyl, fluoro, bromo, and chloro substituents without compromising yield, thereby enabling the rapid synthesis of a wide array of structurally diverse quinazolinone derivatives.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The core of this synthetic breakthrough lies in the intricate interplay between the iron catalyst and the base-promoted activation of the substrates. The reaction mechanism likely initiates with the formation of a carbon-nitrogen bond between the trifluoroethylimidoyl chloride and the isatin nitrogen, facilitated by the presence of sodium hydride. This intermediate then undergoes an iron-catalyzed decarbonylation and subsequent cyclization to form the fused heterocyclic ring system. The use of 4A molecular sieves plays a crucial role in this process by sequestering moisture and potentially stabilizing reactive intermediates, ensuring that the reaction proceeds with high conversion rates even under aerobic conditions. The ability of the ferric chloride catalyst to mediate this transformation under relatively mild thermal conditions (120°C) suggests a low activation energy pathway that is both kinetically favorable and thermodynamically stable.
From an impurity control perspective, the specificity of this iron-catalyzed cycle is advantageous for maintaining high product purity. Unlike radical-based fluorination methods that can lead to indiscriminate fluorination patterns, this cyclization approach directs the trifluoromethyl group specifically to the 2-position of the quinazolinone ring. The robustness of the catalytic system minimizes the formation of side products such as hydrolysis byproducts or oligomers, which are common issues in amide coupling reactions. This inherent selectivity simplifies the downstream purification process, reducing the burden on quality control laboratories and ensuring that the final active pharmaceutical ingredient (API) intermediate meets stringent regulatory specifications for impurity levels. The mechanistic understanding of this pathway allows process chemists to fine-tune reaction parameters, such as stoichiometry and temperature ramps, to further optimize the yield and purity profile for commercial batches.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
The operational simplicity of this synthesis makes it an ideal candidate for technology transfer from laboratory to pilot plant. The procedure involves a straightforward one-pot reaction where all reagents are combined in a polar aprotic solvent, typically DMF, which effectively solubilizes both the organic substrates and the inorganic catalyst. The reaction profile involves an initial incubation period at a lower temperature followed by heating to drive the cyclization to completion. This two-stage temperature protocol helps to manage the exothermic nature of the initial bond formation while ensuring full conversion during the ring-closing step. For detailed standard operating procedures and specific stoichiometric ratios validated through extensive experimentation, please refer to the standardized synthesis guide below.
- Combine ferric chloride, sodium hydride, 4A molecular sieves, trifluoroethylimidoyl chloride, and isatin in an organic solvent such as DMF.
- Stir the mixture at 40°C for 8-10 hours, then increase temperature to 120°C and react for 18-20 hours under air.
- Filter the reaction mixture, mix with silica gel, and purify via column chromatography to isolate the final quinazolinone compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible strategic benefits that extend beyond mere chemical elegance. The primary advantage lies in the substantial reduction of raw material costs driven by the substitution of expensive catalysts and specialized reagents with commodity chemicals. Ferric chloride is a ubiquitous industrial chemical with a stable global supply, insulating manufacturers from the price volatility often associated with precious metals like palladium or platinum. Additionally, the use of isatin and aromatic amines as starting materials leverages existing supply chains for bulk fine chemicals, ensuring consistent availability and reducing the risk of production delays due to raw material shortages. This reliability is paramount for maintaining continuous manufacturing operations and meeting the just-in-time delivery requirements of major pharmaceutical clients.
- Cost Reduction in Manufacturing: The elimination of precious metal catalysts removes the need for costly and complex metal scavenging steps during purification, which significantly lowers processing expenses. Furthermore, the high atom economy of the cyclization reaction minimizes waste generation, reducing the costs associated with solvent recovery and waste disposal. The overall process efficiency translates to a lower cost of goods sold (COGS), providing a competitive pricing advantage in the global market for pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: By relying on widely available starting materials such as isatin and substituted anilines, the supply chain becomes more resilient to disruptions. These commodities are produced by multiple vendors worldwide, preventing single-source dependency risks. The robustness of the reaction conditions, which tolerate air and moisture to a reasonable extent due to the use of molecular sieves, also reduces the need for specialized inert atmosphere equipment, simplifying the manufacturing infrastructure requirements.
- Scalability and Environmental Compliance: The protocol has been demonstrated to be scalable, with the potential to transition from gram-scale laboratory synthesis to multi-kilogram commercial production without significant re-engineering. The use of iron, a non-toxic and environmentally benign metal, aligns with increasingly strict environmental regulations regarding heavy metal discharge. This eco-friendly profile facilitates easier regulatory approval and supports the sustainability goals of modern pharmaceutical companies, enhancing the marketability of the final drug product.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis method. These insights are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing a clear understanding of the process capabilities and limitations. Understanding these details is essential for R&D teams evaluating the feasibility of this route for their specific pipeline projects and for procurement teams assessing the long-term viability of the supply source.
Q: What are the advantages of using FeCl3 over precious metal catalysts for quinazolinone synthesis?
A: Using ferric chloride significantly reduces raw material costs compared to palladium or rhodium catalysts. It also simplifies the purification process by eliminating the need for rigorous heavy metal removal steps, which is critical for pharmaceutical compliance.
Q: What is the substrate scope for this trifluoromethyl quinazolinone synthesis?
A: The method demonstrates excellent functional group tolerance, accommodating various substituents on the aryl ring including methyl, fluoro, bromo, chloro, and methoxy groups, allowing for the synthesis of diverse analogues.
Q: Is this process suitable for industrial scale-up?
A: Yes, the protocol utilizes commercially available reagents like isatin and operates under relatively mild conditions (DMF solvent, air atmosphere), making it highly amenable to scaling from gram-level laboratory synthesis to multi-kilogram production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the success of drug development programs. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and reliability. We are committed to delivering 2-trifluoromethyl quinazolinones with stringent purity specifications, supported by our rigorous QC labs that employ state-of-the-art analytical instrumentation to verify every batch. Our dedication to quality assurance guarantees that the materials you receive are fully compliant with international regulatory standards, facilitating a smooth transition through clinical trials and into commercial manufacturing.
We invite you to collaborate with us to leverage this advanced synthetic technology for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our optimized manufacturing processes can accelerate your timeline and reduce your overall development costs. Let us be your partner in bringing innovative therapies to market faster and more efficiently.