Advanced Asymmetric [3+2] Cyclization for Commercial Scale-up of Complex Chiral Isonucleoside Analogs
The pharmaceutical industry continuously seeks robust synthetic routes for nucleoside analogs, particularly those with enhanced metabolic stability against hydrolysis and enzymatic degradation. Patent CN110642843A introduces a groundbreaking methodology for synthesizing chiral isonucleoside analogs through an asymmetric [3+2] cyclization reaction, representing a significant leap forward in organic synthesis technology. Unlike natural nucleosides which possess vulnerable aminal structures, isonucleosides feature a shifted base configuration that confers superior stability, making them critical candidates for anti-HIV and antitumor therapies. This patent discloses a catalytic system that bypasses the traditional reliance on stoichiometric chiral sources, utilizing instead a palladium catalyst in conjunction with a chiral ferrocene oxazoline derivative nitrogen phosphine ligand. The result is a highly efficient transformation that delivers chiral heteronucleoside analogues with exceptional enantioselectivity reaching up to 98% ee and diastereoselectivity ratios as high as 19:1. For R&D directors and procurement specialists, this technology signals a shift towards more sustainable and cost-effective manufacturing paradigms for complex pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of chiral isonucleosides has been plagued by inefficient synthetic strategies that hinder commercial scalability and economic viability. Traditional pathways typically involve the meticulous design and multi-step synthesis of a chiral tetrahydrofuran ring possessing specific stereoconfiguration and functional groups, which must subsequently be chemically linked to a nucleobase. Alternatively, some methods introduce an amino group onto a pre-existing chiral ring to build the base structure. Both approaches suffer from a critical dependency on equivalent amounts of expensive chiral starting materials, often derived from the chiral pool, which drastically inflates raw material costs. Furthermore, these multi-step sequences invariably lead to cumulative yield losses, resulting in very low overall efficiency. The difficulty in preparing these specialized chiral substrates, combined with the rigorous purification requirements needed to remove impurities generated at each step, creates a bottleneck for supply chain continuity. Consequently, manufacturers face significant challenges in securing reliable supplies of high-purity isonucleoside intermediates at a price point suitable for widespread therapeutic application.
The Novel Approach
In stark contrast to these legacy methods, the technology disclosed in patent CN110642843A employs a direct asymmetric [3+2] cyclization strategy that fundamentally reshapes the production landscape. By utilizing nitrogen-containing heterocyclic substituted olefins and epoxybutene as readily available achiral raw materials, this method eliminates the need for pre-formed chiral centers in the starting feedstock. The reaction proceeds in the presence of a palladium catalyst and a specifically designed chiral ligand, facilitating the simultaneous formation of the ring structure and the installation of chirality in a single operational step. This convergence not only simplifies the synthetic route but also dramatically improves atom economy and overall yield. The ability to derive various functional group-substituted chiral heteronucleosides from the core intermediate with high efficiency further underscores the versatility of this platform. For procurement managers, this translates to a substantial reduction in the complexity of the supply chain, as the reliance on scarce chiral building blocks is removed, thereby enhancing the security and predictability of raw material sourcing for large-scale manufacturing operations.
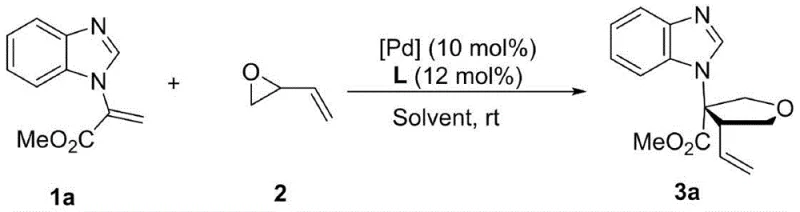
Mechanistic Insights into Pd-Catalyzed Asymmetric [3+2] Cyclization
The core of this technological breakthrough lies in the sophisticated interplay between the palladium catalyst and the chiral ligand environment, which dictates the stereochemical outcome of the cyclization. The reaction mechanism involves the activation of the olefin substrate by the palladium center, followed by a regioselective and stereoselective attack on the vinyl epoxide. The use of ferrocene-derived nitrogen phosphine ligands, particularly the L10 ligand identified in the optimization studies, creates a rigid chiral pocket that effectively differentiates between the enantiotopic faces of the reacting species. This precise spatial arrangement ensures that the newly formed stereocenters are established with high fidelity, minimizing the formation of unwanted diastereomers. The patent data indicates that the choice of ligand is paramount, with L10 outperforming other bisphosphine and monophosphine variants in terms of both yield and enantiomeric excess. Understanding this mechanistic nuance is vital for R&D teams aiming to replicate or adapt this chemistry for specific analog targets, as slight modifications to the ligand structure or the electronic properties of the olefin can significantly influence the reaction trajectory and final product purity.
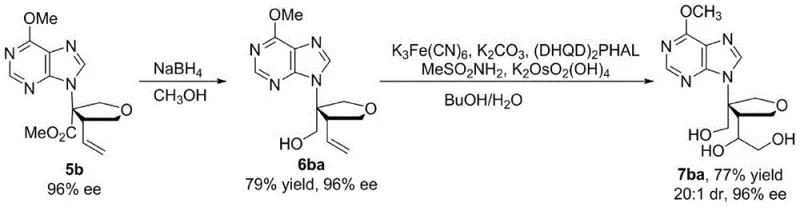
Beyond the primary cyclization event, the control of impurity profiles is a critical aspect of this synthesis that directly impacts downstream processing and regulatory compliance. The high diastereoselectivity observed, with dr values reaching 19:1, implies that the formation of side products with incorrect relative stereochemistry is effectively suppressed. This inherent selectivity reduces the burden on purification processes such as chromatography or crystallization, which are often the most costly and time-consuming stages in pharmaceutical manufacturing. Furthermore, the compatibility of the reaction conditions with various functional groups on the nitrogen heterocycle allows for the direct synthesis of diverse analogs without extensive protecting group manipulations. The subsequent derivatization of the core isonucleoside scaffold, as demonstrated by the conversion to compounds bearing multiple hydroxyl groups or halogen substituents, proceeds with retention of stereochemical integrity. This robustness ensures that the optical purity established during the initial cyclization is maintained throughout the synthetic sequence, delivering final active pharmaceutical ingredients that meet stringent quality specifications required by global health authorities.
How to Synthesize Chiral Isonucleoside Analogs Efficiently
The implementation of this asymmetric [3+2] cyclization protocol requires careful attention to reaction parameters to maximize yield and stereoselectivity. The process begins with the preparation of the catalytic system, where a palladium source such as Pd(PPh3)4 is combined with the chiral ligand L10 in an appropriate organic solvent like dichloromethane. The substrate, typically an alpha-heterocyclic substituted acrylate, is then introduced along with the vinyl epoxide partner under an inert nitrogen atmosphere to prevent catalyst deactivation. Maintaining the reaction temperature within the optimal range of -40°C to 60°C is crucial for balancing reaction rate and stereocontrol. Detailed standardized synthesis steps see the guide below.
- Prepare the reaction mixture by combining nitrogen-containing heterocyclic substituted olefin and epoxybutene with a palladium catalyst such as Pd(PPh3)4 and a chiral ferrocene-derived nitrogen phosphine ligand like L10 in an organic solvent.
- Maintain the reaction temperature between -40°C and 60°C under a nitrogen atmosphere, allowing the asymmetric [3+2] cyclization to proceed until the formation of the chiral isonucleoside analog intermediate is complete.
- Isolate the target product through standard workup procedures including extraction and column chromatography, achieving high enantioselectivity up to 98% ee and excellent diastereoselectivity.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this patented synthesis method offers profound advantages that extend well beyond the laboratory bench, addressing key pain points for procurement and supply chain leadership. The most significant benefit is the drastic simplification of the raw material portfolio; by shifting from expensive, scarce chiral starting materials to abundant achiral olefins and epoxides, manufacturers can achieve substantial cost savings in chemical procurement. This transition mitigates the risk of supply disruptions associated with niche chiral intermediates and allows for more accurate long-term budget forecasting. Additionally, the reduction in synthetic steps directly correlates with lower operational expenditures, as fewer unit operations mean reduced consumption of solvents, energy, and labor hours. The high efficiency of the catalytic system also minimizes waste generation, aligning with increasingly strict environmental regulations and sustainability goals that modern chemical enterprises must uphold to maintain their social license to operate.
- Cost Reduction in Manufacturing: The elimination of stoichiometric chiral auxiliaries and the reduction of synthetic steps fundamentally alter the cost structure of isonucleoside production. Traditional routes often incur high costs due to the purchase of premium chiral building blocks and the losses associated with multi-step purification; this new method circumvents those expenses by generating chirality catalytically. The high yields reported, often exceeding 90% for the key cyclization step, ensure that raw material utilization is maximized, further driving down the cost per kilogram of the final intermediate. Moreover, the use of common solvents and standard palladium catalysts, which can potentially be recovered and recycled, adds another layer of economic efficiency. These factors combine to create a manufacturing process that is not only technically superior but also financially compelling for large-scale commercial ventures.
- Enhanced Supply Chain Reliability: Supply chain resilience is significantly bolstered by the reliance on commodity chemicals rather than specialized chiral reagents. The starting materials, such as substituted acrylates and epoxybutenes, are widely available from multiple global suppliers, reducing the risk of single-source dependency. This diversification of the supply base ensures continuity of production even in the face of market volatility or logistical challenges affecting specific regions. Furthermore, the robustness of the reaction conditions, which tolerate a range of temperatures and solvents, provides operational flexibility that allows manufacturing sites to adapt to local resource availability. For supply chain heads, this means a more predictable lead time for high-purity pharmaceutical intermediates and the ability to scale production volumes rapidly in response to market demand without being constrained by raw material bottlenecks.
- Scalability and Environmental Compliance: The scalability of this process is evidenced by its mild reaction conditions and the absence of hazardous reagents that typically complicate scale-up efforts. Operating at near-ambient temperatures and pressures reduces the engineering controls required for safe manufacturing, facilitating a smoother transition from pilot plant to commercial production. The high atom economy of the [3+2] cycloaddition minimizes the generation of by-product waste, simplifying effluent treatment and reducing the environmental footprint of the facility. This alignment with green chemistry principles is increasingly important for meeting corporate sustainability targets and regulatory compliance standards. The ability to produce complex chiral molecules with minimal waste and energy input positions this technology as a future-proof solution for the sustainable manufacturing of next-generation antiviral and anticancer therapeutics.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this asymmetric synthesis technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on the practical application of the method. Understanding these details is essential for stakeholders evaluating the feasibility of integrating this route into their existing manufacturing portfolios. The answers reflect the specific advantages in terms of selectivity,原料 availability, and process robustness that distinguish this approach from conventional methodologies.
Q: What are the primary advantages of this asymmetric [3+2] cyclization method over traditional nucleoside synthesis?
A: Traditional methods often require expensive chiral starting materials and multi-step sequences to construct the tetrahydrofuran ring, resulting in low overall yields. This patented approach utilizes readily available achiral raw materials and a single catalytic step to achieve high stereoselectivity (up to 98% ee), significantly simplifying the process and reducing material costs.
Q: How does the choice of ligand impact the enantioselectivity in this palladium-catalyzed reaction?
A: The patent highlights that ferrocene-derived oxazoline ligands, specifically L10, provide superior stereocontrol compared to other bisphosphine or nitrogen-phosphine ligands. The unique steric and electronic properties of the L10 ligand facilitate the formation of the desired chiral center with a diastereomeric ratio of up to 19:1, ensuring high optical purity essential for pharmaceutical applications.
Q: Can this synthetic route be adapted for large-scale commercial production of antiviral intermediates?
A: Yes, the reaction conditions are mild, operating between -40°C and 60°C in common organic solvents like dichloromethane or toluene. The use of robust palladium catalysts and the elimination of sensitive chiral pools make the process highly scalable, addressing key supply chain concerns regarding continuity and cost-efficiency for bulk manufacturing of isonucleoside derivatives.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral Isonucleoside Analogs Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced catalytic technologies like the asymmetric [3+2] cyclization described in patent CN110642843A for the future of nucleoside drug development. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative laboratory discoveries are successfully translated into reliable industrial realities. Our commitment to quality is underpinned by stringent purity specifications and rigorous QC labs that utilize state-of-the-art analytical instrumentation to verify every batch. We understand that for R&D directors, the consistency of the impurity profile is just as critical as the yield, and our process engineering teams are dedicated to optimizing these parameters to meet the exacting standards of the global pharmaceutical industry.
We invite you to collaborate with us to leverage this cutting-edge synthesis route for your specific project needs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis that quantifies the economic benefits of switching to this catalytic method for your supply chain. Please contact us to request specific COA data and route feasibility assessments tailored to your target molecules. By partnering with NINGBO INNO PHARMCHEM, you gain access to a wealth of chemical expertise and manufacturing capacity designed to accelerate your time-to-market while optimizing your production costs.