Advanced Palladium-Catalyzed Asymmetric [3+2] Cyclization for Commercial Scale Chiral Isonucleosides
Introduction to Next-Generation Isonucleoside Synthesis
The landscape of antiviral drug development is constantly evolving, driven by the need for more stable and potent nucleoside analogs. Natural nucleoside drugs often suffer from metabolic instability due to their aminal structures, which are prone to hydrolysis and enzymatic degradation. To address this critical limitation, the pharmaceutical industry has shifted focus toward isonucleosides, where the base is shifted to enhance stability while retaining biological activity. A groundbreaking advancement in this field is detailed in patent CN110642843A, which discloses a highly efficient method for synthesizing chiral isonucleoside analogs via an asymmetric [3+2] cyclization reaction. This technology represents a paradigm shift from traditional multi-step syntheses, offering a direct, convergent route to complex chiral heterocycles. By leveraging a palladium-catalyzed system with specialized chiral ligands, this method achieves unprecedented levels of stereocontrol, making it a vital asset for any organization seeking a reliable pharmaceutical intermediate supplier capable of delivering high-value antiviral scaffolds.
The core innovation lies in the ability to construct the chiral tetrahydrofuran ring directly from achiral starting materials. Specifically, the reaction couples nitrogen heterocyclic substituted olefins with vinyl epoxides (epoxybutene) in the presence of a palladium catalyst and a chiral ferrocene oxazoline derivative. This approach bypasses the need for pre-existing chiral centers in the starting materials, which are often costly and difficult to source. The result is a streamlined process that delivers chiral isonucleoside analogs with yields reaching up to 93% and enantioselectivity as high as 98% ee. For R&D teams focused on cost reduction in API manufacturing, this methodology offers a compelling alternative to legacy routes, promising significant improvements in both process efficiency and final product purity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of chiral isonucleosides has been fraught with synthetic challenges that hinder commercial viability. Traditional pathways typically rely on one of two strategies: either designing a chiral tetrahydrofuran ring through a lengthy multi-step sequence followed by coupling with a nucleobase, or introducing an amino group onto a pre-formed chiral ring to build the base. Both approaches share a critical flaw: they are dependent on stoichiometric amounts of chiral sources. These chiral starting materials are often derived from the "chiral pool," which can be expensive, subject to supply chain volatility, and limited in structural diversity. Furthermore, the multi-step nature of these conventional routes inevitably leads to cumulative yield losses, where the overall efficiency drops precipitously with each additional transformation. The difficulty in preparing these specific chiral substrates, combined with low overall yields, creates a significant bottleneck in the supply chain for high-purity chiral isonucleoside analogs, driving up costs and extending lead times for drug development programs.
The Novel Approach
In stark contrast, the method described in CN110642843A introduces a catalytic asymmetric [3+2] cycloaddition that fundamentally reimagines the bond construction strategy. Instead of relying on expensive chiral building blocks, this novel approach utilizes cheap, readily available achiral raw materials—specifically α-heterocyclic substituted acrylates and vinyl epoxides. The magic of this transformation occurs through the precise orchestration of a palladium catalyst and a chiral ligand, which induces asymmetry during the ring-forming event itself. This catalytic turnover means that a small amount of chiral information (the ligand) can generate a large amount of chiral product, drastically improving atom economy. The reaction tolerates a wide range of nitrogen heterocycles, including purines, benzotriazoles, and various substituted imidazoles, demonstrating remarkable versatility. By eliminating the dependency on stoichiometric chiral auxiliaries and reducing the step count, this method provides a simple, convenient, and highly efficient pathway that is ideally suited for the commercial scale-up of complex heterocyclic compounds.
Mechanistic Insights into Pd-Catalyzed Asymmetric [3+2] Cyclization
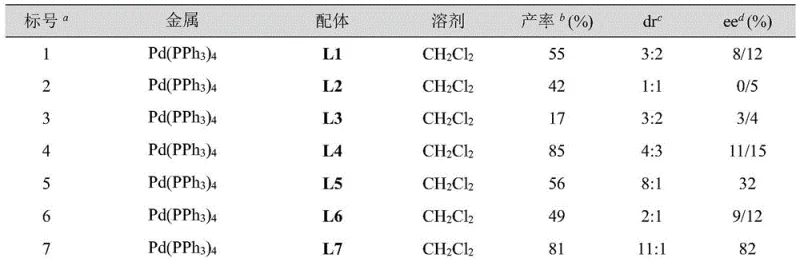
To fully appreciate the technical sophistication of this process, one must delve into the mechanistic nuances of the palladium-catalyzed cycle. The reaction employs a zero-valent palladium species, typically Pd(PPh3)4, which coordinates with a chiral ferrocene-derived nitrogen-phosphine ligand. Among the library of ligands screened, the ferrocenyl oxazoline derivatives, particularly ligand L10, exhibited superior performance. The steric bulk and electronic properties of the ferrocene backbone create a highly defined chiral pocket around the metal center. When the electron-deficient olefin (the acrylate derivative) and the vinyl epoxide coordinate to this chiral palladium complex, the subsequent oxidative addition and migratory insertion steps are strictly controlled. This ensures that the new carbon-carbon and carbon-oxygen bonds are formed with specific spatial orientation, leading to the observed high diastereoselectivity (up to 19:1 dr) and enantioselectivity (up to 98% ee). The ability to activate electron-deficient olefins that possess both electron-donating amine groups and electron-withdrawing ester groups—a combination previously considered chemically inert in this context—is a testament to the unique reactivity profile of this catalytic system.
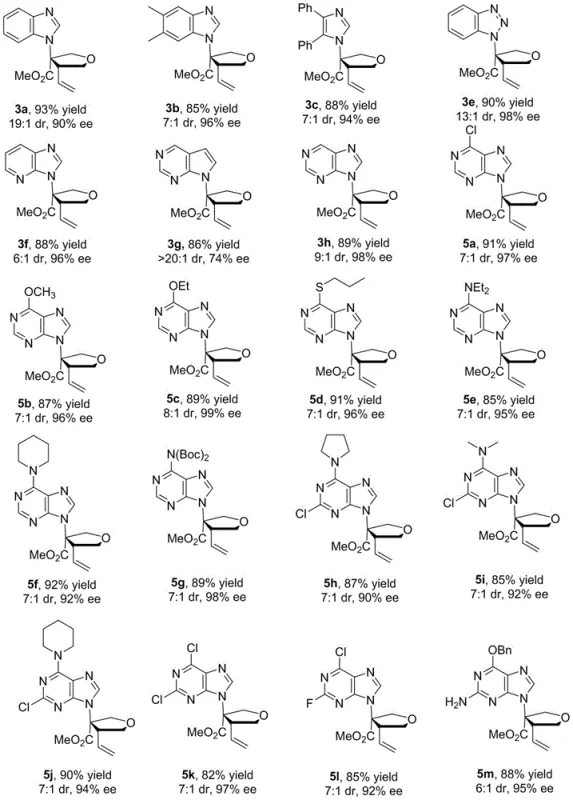
Beyond the primary cyclization, the robustness of the resulting chiral isonucleoside scaffold allows for extensive downstream functionalization, which is critical for structure-activity relationship (SAR) studies. The patent details how the initial cyclization product (compound 3) can be further derivatized through reduction, bromination, dihydroxylation, and fluorination to yield a diverse array of functionalized analogs (compounds 6-11). For instance, reduction with sodium borohydride yields mono-hydroxylated derivatives, while dihydroxylation using potassium ferricyanide and osmium tetroxide generates tri-hydroxylated species. This modularity implies that a single optimized process can serve as a platform for generating entire libraries of potential drug candidates. From an impurity control perspective, the high stereoselectivity of the initial step minimizes the formation of unwanted diastereomers, simplifying the purification process and ensuring that the final high-purity chiral isonucleoside analogs meet stringent regulatory specifications without the need for cumbersome chiral resolution steps.
How to Synthesize Chiral Isonucleoside Analogs Efficiently
Implementing this advanced synthetic route requires careful attention to reaction parameters to maximize yield and selectivity. The process is designed to be operationally simple, typically conducted in common organic solvents such as dichloromethane at room temperature. The molar ratios of the substrates, catalyst, and ligand are optimized to balance reaction rate with cost efficiency, generally employing a slight excess of the epoxybutene relative to the olefin substrate. While the specific laboratory protocols involve precise weighing and inert atmosphere techniques, the fundamental steps are straightforward and scalable. For detailed operational procedures, safety data, and specific purification parameters tailored to your specific substrate, please refer to the standardized synthesis guide below.
- Prepare the reaction mixture by combining the nitrogen-containing heterocyclic substituted olefin and epoxybutene with a palladium catalyst such as Pd(PPh3)4 and a chiral ferrocene-derived oxazoline ligand like L10 in an organic solvent.
- Maintain the reaction under a nitrogen atmosphere at room temperature for approximately 24 hours to allow the asymmetric [3+2] cyclization to proceed to completion.
- Quench the reaction, perform aqueous workup with dichloromethane and water, dry the organic phase, and purify the resulting chiral isonucleoside analog via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this catalytic asymmetric synthesis offers tangible strategic benefits that extend beyond mere technical elegance. The primary advantage lies in the radical simplification of the raw material supply chain. By shifting from complex, multi-step chiral precursors to commodity chemicals like acrylates and vinyl epoxides, manufacturers can mitigate the risks associated with sourcing specialized chiral building blocks. This substitution not only stabilizes the supply chain but also drives down the baseline cost of goods sold (COGS). Furthermore, the high efficiency of the catalytic system means that less material is wasted in side reactions or lost during purification, contributing to a more sustainable and cost-effective manufacturing process. The ability to produce diverse analogs from a common intermediate also enhances supply chain agility, allowing for rapid response to changing market demands for specific antiviral candidates.
- Cost Reduction in Manufacturing: The elimination of stoichiometric chiral auxiliaries and the reduction in synthetic step count directly translate to significant cost savings. Traditional routes often require expensive resolving agents or chiral pool materials that dominate the cost structure; this new method replaces those high-cost inputs with a catalytic amount of ligand and inexpensive achiral feedstocks. Additionally, the high yields and selectivity reduce the burden on downstream purification, lowering solvent consumption and waste disposal costs. This holistic improvement in process mass intensity (PMI) ensures a leaner, more economical production model for pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: Reliance on niche chiral starting materials often introduces single points of failure in the supply chain. By utilizing widely available industrial chemicals as starting materials, this process diversifies the supplier base and reduces lead times. The robustness of the reaction conditions—operating effectively at room temperature in standard solvents—further ensures that production is not susceptible to the complexities of cryogenic cooling or high-pressure equipment, facilitating smoother logistics and more predictable delivery schedules for reliable pharmaceutical intermediate supplier networks.
- Scalability and Environmental Compliance: The simplicity of the workup procedure, involving standard extraction and chromatography, makes this process highly amenable to scale-up from kilogram to tonne quantities. The use of dichloromethane, while requiring proper handling, is a well-understood solvent in industrial settings with established recovery protocols. Moreover, the high atom economy of the [3+2] cycloaddition minimizes the generation of byproduct waste, aligning with modern green chemistry principles. This facilitates easier regulatory approval and environmental compliance, reducing the administrative burden on manufacturing sites aiming for cost reduction in API manufacturing.
Frequently Asked Questions (FAQ)
Understanding the technical and commercial implications of this new synthesis method is crucial for stakeholders evaluating its adoption. The following questions address common inquiries regarding the scope, selectivity, and practical application of this technology. These answers are derived directly from the experimental data and beneficial effects reported in the patent literature, providing a factual basis for decision-making regarding the integration of this route into your existing manufacturing portfolio.
Q: What are the primary advantages of this asymmetric [3+2] cyclization method over traditional nucleoside synthesis?
A: Traditional methods often rely on expensive chiral pool precursors and multi-step sequences that suffer from low overall yields. This novel approach utilizes readily available achiral raw materials to directly construct the chiral tetrahydrofuran core with high stereocontrol, significantly simplifying the synthetic route and reducing material costs.
Q: What level of stereoselectivity can be achieved with the L10 ligand system?
A: The optimized protocol utilizing the ferrocene-derived oxazoline ligand L10 demonstrates exceptional stereocontrol, achieving enantiomeric excess (ee) values up to 98% and diastereomeric ratios (dr) as high as 19:1, ensuring the production of high-purity optical isomers required for pharmaceutical applications.
Q: Is this process suitable for large-scale manufacturing of antiviral intermediates?
A: Yes, the reaction operates under mild conditions (room temperature) using common solvents like dichloromethane and standard palladium catalysts. The robustness of the catalytic system and the availability of substrates make it highly amenable to commercial scale-up for producing complex heterocyclic pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral Isonucleoside Analogs Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful translation of innovative academic research into commercial reality requires more than just a patent; it demands deep process engineering expertise and a commitment to quality. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle sensitive organometallic catalysis, ensuring that the delicate balance of the palladium-ligand system is maintained even at large scales. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of chiral isonucleoside analogs meets the exacting standards required for clinical and commercial pharmaceutical applications. Our team is ready to assist you in navigating the complexities of this synthesis, from initial feasibility studies to full-scale GMP manufacturing.
We invite you to explore how this advanced asymmetric cyclization technology can optimize your drug development pipeline. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific project needs. We are prepared to provide specific COA data and comprehensive route feasibility assessments to demonstrate how partnering with us can accelerate your timeline to market while maximizing value. Let us be your trusted partner in bringing the next generation of antiviral therapeutics to the world.