Advanced Synthesis and Purification of Parecoxib Sodium for Global Pharmaceutical Supply Chains
Introduction to Advanced Parecoxib Sodium Manufacturing
The pharmaceutical landscape for postoperative pain management relies heavily on the efficient production of COX-2 inhibitors, with Parecoxib Sodium standing as a critical active pharmaceutical ingredient. Patent CN113845488A introduces a groundbreaking preparation and refining method that addresses long-standing challenges in the synthesis of Parecoxib and its key intermediates. This technology offers a streamlined pathway that significantly enhances production efficiency, yield, and final product purity, making it an essential reference for any reliable pharmaceutical intermediates supplier aiming to optimize their portfolio. The core innovation lies in a meticulously engineered sequence of sulfonation, amination, and acylation steps that bypass the cumbersome purification techniques of the past.
Traditionally, the synthesis of such complex heterocyclic compounds has been plagued by issues related to emulsification during workup, difficult-to-remove isomeric impurities, and the reliance on costly chromatographic purification. The disclosed method overcomes these barriers by introducing specific solvent systems, particularly the strategic use of hydrous ethanol for recrystallization. This approach not only simplifies the operational workflow but also ensures that the final API meets stringent regulatory standards for related substances. By integrating this patented methodology, manufacturers can achieve a level of process robustness that is vital for maintaining supply continuity in the global market.
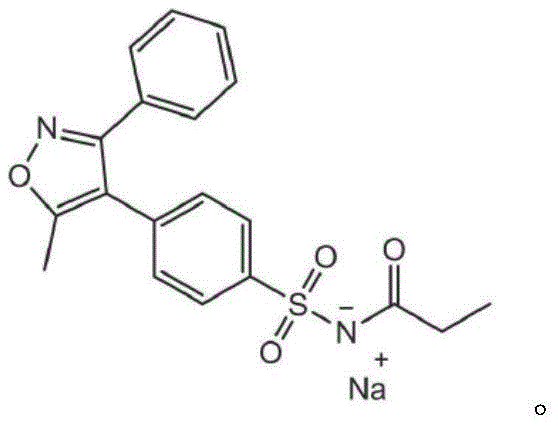
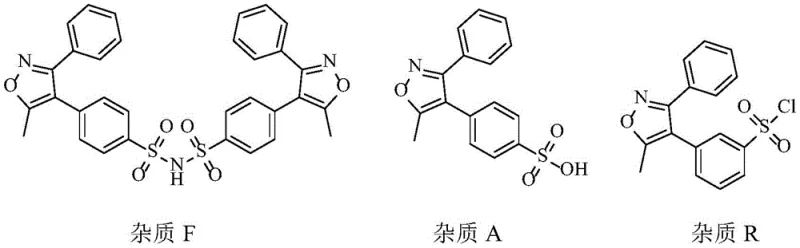
Furthermore, the patent details a comprehensive strategy for impurity management, identifying and controlling specific degradation products such as Impurity F, Impurity A, and Impurity R. This level of detail is crucial for R&D teams focused on cost reduction in API manufacturing, as it reduces the risk of batch failures and reprocessing. The ability to produce high-purity Parecoxib Sodium through a scalable, non-chromatographic route represents a significant leap forward in process chemistry, aligning perfectly with the needs of modern generic drug development and commercial supply chains.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methods, such as those disclosed in WO2003029230A, have historically relied on harsh reaction conditions and inefficient purification protocols that hinder industrial viability. A primary drawback of these conventional routes is the use of trifluoroacetic acid (TFA) during the sulfonation reaction, a reagent known for its strong corrosivity and the safety hazards it poses in large-scale reactors. Moreover, the post-treatment processes in these older methods are notoriously complicated, often requiring repeated treatments with toluene and water, followed by the addition of concentrated ammonia hydroxide. This multi-step workup not only consumes excessive time and labor but also generates significant volumes of hazardous waste.
Perhaps the most significant bottleneck in traditional synthesis is the reliance on column chromatography for purification, as seen in optimizations like WO2005123701A. While chromatography can achieve high purity on a laboratory scale, it is economically and logistically prohibitive for commercial scale-up of complex pharmaceutical intermediates. The low total yield, reported around 25.5% over three steps in some prior methods, combined with the high cost of silica gel and solvents, renders these processes uncompetitive for mass production. Additionally, the use of methanol/water mixtures for recrystallization in these legacy processes often fails to adequately remove specific isomeric impurities, necessitating further refining steps that erode profit margins.
The Novel Approach
In stark contrast, the novel approach detailed in CN113845488A presents a cohesive and industrially friendly synthesis route that eliminates the need for column chromatography entirely. The process begins with a refined sulfonation step where the reaction mixture is quenched and subjected to a liquid separation followed by ethyl acetate extraction. This specific modification effectively prevents the emulsification phenomenon that frequently stalls production lines, thereby greatly shortening working hours and saving solvent consumption. The subsequent recrystallization using n-hexane ensures the removal of isomer Impurity R, setting a high-purity foundation for downstream steps.
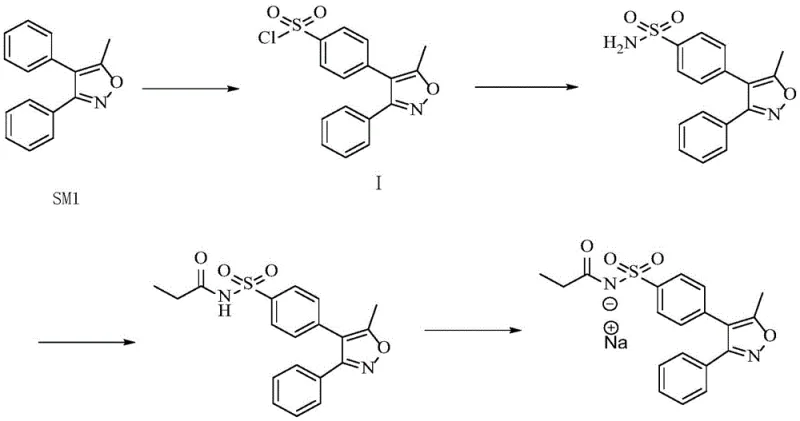
The innovation continues into the amination and acylation stages, where the patent specifies the use of hydrous ethanol with controlled water content (2-20%) for recrystallization. This seemingly simple adjustment leverages the differential solubility of the target molecule versus its impurities, allowing for the effective removal of Impurity F and other by-products without the need for exotic reagents. The entire sequence, from the initial sulfonation of SM1 to the final salification with sodium hydroxide, is designed for reducing lead time for high-purity pharmaceutical intermediates. By replacing corrosive TFA with optimized chlorosulfonic acid protocols and substituting chromatography with crystallization, this method offers a direct path to high-purity Parecoxib Sodium that is both economically superior and environmentally more sustainable.
Mechanistic Insights into Impurity Control and Crystallization Dynamics
From a mechanistic perspective, the success of this synthesis hinges on the precise control of solvation and crystallization thermodynamics during the refining stages. In the sulfonation step, the formation of the sulfonyl chloride intermediate (Intermediate I) is prone to generating isomeric by-products, specifically Impurity R, which arises from substitution at the ortho-position relative to the isoxazole ring. The patent elucidates that the use of n-hexane as an anti-solvent during the recrystallization of Intermediate I selectively precipitates the desired para-isomer while keeping the ortho-isomer in solution. This selectivity is critical because Impurity R can carry through subsequent steps, complicating the final purification of the API.
Moving to the amination step, the conversion of the sulfonyl chloride to the sulfonamide (Valdecoxib) introduces the risk of forming dimeric or oligomeric impurities, collectively referred to as Impurity F. The patent data demonstrates that the water content in the ethanol recrystallization solvent plays a pivotal role in suppressing this impurity. At a water content of approximately 5%, the solubility profile shifts such that Impurity F remains in the mother liquor while Valdecoxib crystallizes out with high purity. This phenomenon suggests a specific interaction between the water molecules and the polar sulfonamide groups, which alters the lattice energy of the crystallizing solid, favoring the exclusion of structurally similar contaminants.
Finally, in the acylation step where Valdecoxib is converted to Parecoxib, the control of temperature and solvent composition is paramount. The reaction with propionic anhydride can generate unreacted starting material and over-acylated by-products. The patented refining process utilizes a pulping technique with aqueous ethanol at 40-45°C, followed by cooling to 0-10°C. This thermal cycling ensures that the kinetic product (Parecoxib) is stabilized while soluble impurities are washed away. The data indicates that temperatures exceeding 45°C during refining can lead to an increase in Impurity I, highlighting the narrow operational window required for optimal quality. Understanding these mechanistic nuances allows process chemists to fine-tune parameters for maximum yield and minimal waste, ensuring the production of high-purity Parecoxib Sodium suitable for sensitive parenteral formulations.
How to Synthesize Parecoxib Efficiently
The synthesis of Parecoxib via this patented route involves a logical progression of four distinct chemical transformations, each optimized for scalability and purity. The process begins with the sulfonation of the isoxazole precursor, followed by amination to form the sulfonamide core, acylation to introduce the propionyl group, and finally salification to generate the sodium salt. Each step incorporates specific workup procedures, such as ethyl acetate extraction and hydrous ethanol recrystallization, which are critical for removing the specific impurities identified in the patent. For a detailed breakdown of the reaction conditions, stoichiometry, and operational parameters required to replicate this high-efficiency route, please refer to the standardized synthesis guide below.
- Perform sulfonation of SM1 with chlorosulfonic acid in dichloromethane, followed by ethyl acetate extraction and n-hexane recrystallization to obtain high-purity Intermediate I.
- Conduct amination of Intermediate I using ammonia water, followed by recrystallization in hydrous ethanol (5-10% water) to remove Impurity F and yield Valdecoxib.
- Execute acylation of Valdecoxib with propionic anhydride in THF, refining the crude product using aqueous ethanol at 40-45°C to achieve final Parecoxib purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis method translates into tangible strategic advantages that go beyond mere technical specifications. The elimination of column chromatography is perhaps the most significant cost driver, as it removes the need for expensive silica gel, vast quantities of elution solvents, and the associated labor for packing and running columns. This simplification drastically reduces the variable cost per kilogram of the API, allowing for more competitive pricing in tender situations. Furthermore, the reduction in processing time—achieved by avoiding emulsification and streamlining extractions—means that reactor turnover rates can be increased, effectively boosting plant capacity without capital expenditure on new equipment.
- Cost Reduction in Manufacturing: The process achieves substantial cost savings by replacing high-cost purification methods with standard crystallization techniques. By utilizing common solvents like ethyl acetate, ethanol, and n-hexane, the method avoids the procurement complexities and price volatility associated with specialized reagents like trifluoroacetic acid or large volumes of chromatography-grade solvents. The improved yield at each stage, particularly the high recovery of Valdecoxib and Parecoxib, ensures that raw material costs are amortized over a larger output of saleable product, directly improving the gross margin profile for manufacturers.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route significantly mitigates the risk of supply disruptions. Traditional methods prone to emulsification often result in unpredictable batch cycles and potential losses of material during phase separation. By engineering a process that separates cleanly and crystallizes reliably, the supply chain becomes more predictable and resilient. This reliability is crucial for meeting the just-in-time delivery requirements of major pharmaceutical clients, ensuring that stockouts are minimized and long-term supply agreements can be honored with confidence.
- Scalability and Environmental Compliance: From an environmental and regulatory standpoint, this method offers a cleaner profile that simplifies waste management. The reduction in solvent usage and the avoidance of corrosive acids lower the burden on wastewater treatment facilities and reduce the generation of hazardous waste streams. This alignment with green chemistry principles not only lowers disposal costs but also facilitates easier regulatory approval in markets with strict environmental standards. The process is inherently designed for commercial scale-up of complex pharmaceutical intermediates, allowing for seamless transition from pilot plant to multi-ton production without the need for fundamental process re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Parecoxib Sodium synthesis method. These insights are derived directly from the experimental data and comparative studies presented in the patent, offering clarity on how this technology outperforms legacy processes. Understanding these specifics is vital for technical teams evaluating the feasibility of adopting this route for their own manufacturing operations or for sourcing partners who utilize it.
Q: How does this patent improve impurity control compared to prior art?
A: The patent introduces specific recrystallization protocols using hydrous ethanol to effectively remove critical impurities like Impurity F and Impurity R, which are difficult to eliminate using conventional column chromatography methods described in earlier patents like WO2003029230A.
Q: What are the key advantages for industrial scale-up?
A: The process eliminates the need for expensive and time-consuming column chromatography, replaces corrosive trifluoroacetic acid with optimized chlorosulfonic acid handling, and utilizes standard solvent systems like ethyl acetate and ethanol, making it highly suitable for multi-ton commercial production.
Q: What is the optimal water content for the refining solvent?
A: For the amination step, 5% water content in ethanol is preferred to minimize Impurity F. For the acylation step, 10% water content in ethanol provides the best balance of yield and purity during the pulping and crystallization phases.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Parecoxib Sodium Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent literature to commercial reality requires deep technical expertise and robust infrastructure. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this patented process are fully realized in practice. Our state-of-the-art facilities are equipped to handle the specific solvent systems and temperature controls required for this synthesis, and our rigorous QC labs enforce stringent purity specifications to guarantee that every batch of Parecoxib Sodium meets the highest global pharmacopoeial standards.
We invite potential partners to engage with our technical procurement team to discuss how this advanced manufacturing route can optimize your supply chain. By leveraging our capabilities, you can secure a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating exactly how this efficient synthesis translates into bottom-line value. We encourage you to contact us today to request specific COA data and route feasibility assessments, and let us demonstrate why we are the preferred choice for reliable pharmaceutical intermediates supplier partnerships in the global market.