Advanced Synthesis of Benzopyran-3-ol Derivatives via Cross-Dehydrogenation Coupling for Commercial Scale-up
The pharmaceutical and fine chemical industries are constantly seeking more efficient pathways to access privileged scaffolds like benzopyran-3-ol derivatives, which serve as critical cores for numerous bioactive compounds ranging from antioxidants to antihypertensives. Patent CN111423405B introduces a groundbreaking methodology that leverages cross-dehydrogenation coupling (CDC) technology to construct these complex architectures directly from simpler precursors. This innovation represents a significant paradigm shift from traditional cross-coupling reactions, offering a streamlined route that bypasses the need for pre-functionalized substrates. For R&D directors and process chemists, this patent outlines a robust protocol that utilizes palladium catalysis to achieve intramolecular C(sp3)-H arylation with remarkable selectivity. The technical depth of this disclosure provides a solid foundation for developing scalable manufacturing processes that address the growing demand for high-purity pharmaceutical intermediates. By integrating this advanced synthetic strategy, manufacturers can potentially overcome longstanding bottlenecks associated with multi-step functionalization sequences.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of C(sp3)-C(sp2) bonds in benzopyran systems has relied heavily on classical methods such as Friedel-Crafts reactions or transition metal-catalyzed cross-couplings like Suzuki-Miyaura and Negishi reactions. While these established techniques are widely understood, they suffer from inherent inefficiencies that complicate large-scale production and increase environmental burdens. Specifically, these conventional routes necessitate the pre-functionalization of both coupling partners, requiring the installation of halides and organometallic reagents which generates substantial stoichiometric waste. Furthermore, Friedel-Crafts methodologies often exhibit poor regioselectivity, particularly with mono-substituted aromatic hydrocarbons, leading to difficult-to-separate isomeric mixtures that compromise final product purity. The reliance on harsh reaction conditions and sensitive reagents in these traditional protocols also poses significant safety challenges during commercial scale-up. Consequently, the industry has long sought a more direct approach that minimizes step count while maximizing atom economy and operational simplicity.
The Novel Approach
The methodology disclosed in the patent revolutionizes this landscape by employing a direct intramolecular C(sp3)-H arylation strategy via cross-dehydrogenation coupling. This novel approach allows for the direct formation of the critical carbon-carbon bond without the prerequisite of pre-installed leaving groups, thereby drastically simplifying the synthetic sequence. 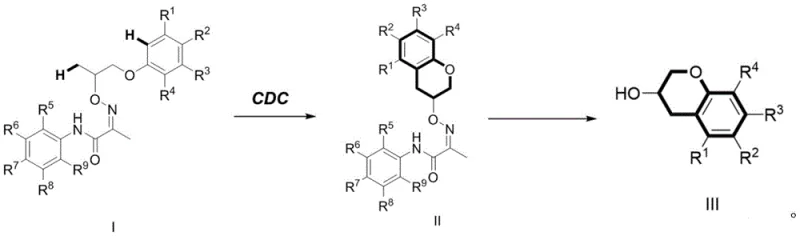 The process initiates with a palladium-catalyzed cyclization of Compound I, utilizing an oxidant to facilitate the C-H activation event under relatively mild thermal conditions. Following the formation of the benzopyran core, a subsequent treatment with molybdenum hexacarbonyl efficiently removes the directing group to yield the final alcohol derivative. This two-step sequence not only improves the overall yield but also enhances the universality of the synthesis across a broad range of substituents. By eliminating the need for separate halogenation and metallation steps, this route offers a compelling solution for cost reduction in pharmaceutical intermediate manufacturing.
The process initiates with a palladium-catalyzed cyclization of Compound I, utilizing an oxidant to facilitate the C-H activation event under relatively mild thermal conditions. Following the formation of the benzopyran core, a subsequent treatment with molybdenum hexacarbonyl efficiently removes the directing group to yield the final alcohol derivative. This two-step sequence not only improves the overall yield but also enhances the universality of the synthesis across a broad range of substituents. By eliminating the need for separate halogenation and metallation steps, this route offers a compelling solution for cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Pd-Catalyzed Intramolecular C-H Arylation
The core of this synthetic breakthrough lies in the sophisticated mechanism of palladium-catalyzed C-H activation, which enables the selective functionalization of inert C-H bonds. The reaction begins with the coordination of the palladium catalyst to the directing group present on Compound I, which serves to orient the metal center in proximity to the target C(sp3)-H bond. 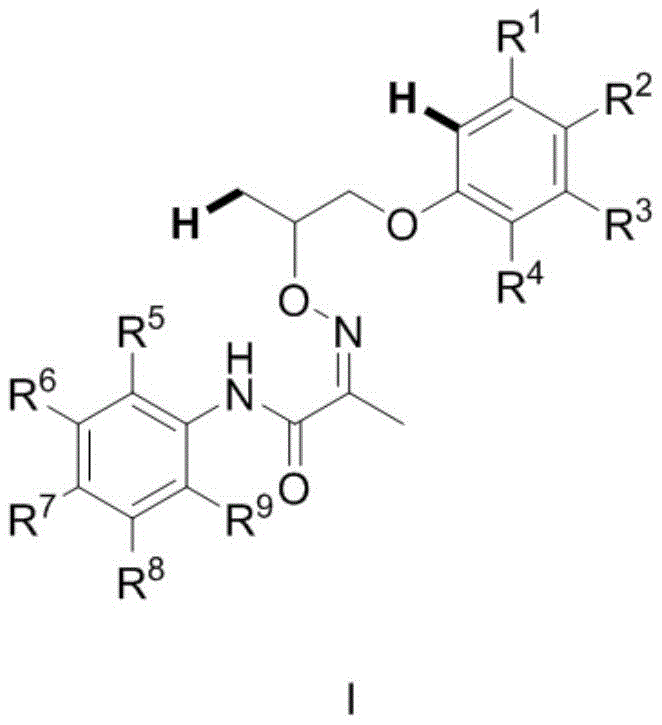 Upon oxidation, the palladium species facilitates the cleavage of the C-H bond and the subsequent formation of the C-C bond with the aromatic ring, closing the pyran ring system. The use of specific oxidants, such as N-fluorobisbenzenesulfonamide, is critical for regenerating the active catalytic species and driving the reaction forward to completion. This mechanistic pathway ensures high regioselectivity, as the directing group effectively controls the site of activation, minimizing the formation of unwanted byproducts. Understanding this catalytic cycle is essential for process optimization, as it allows chemists to fine-tune ligand and oxidant ratios to maximize turnover numbers.
Upon oxidation, the palladium species facilitates the cleavage of the C-H bond and the subsequent formation of the C-C bond with the aromatic ring, closing the pyran ring system. The use of specific oxidants, such as N-fluorobisbenzenesulfonamide, is critical for regenerating the active catalytic species and driving the reaction forward to completion. This mechanistic pathway ensures high regioselectivity, as the directing group effectively controls the site of activation, minimizing the formation of unwanted byproducts. Understanding this catalytic cycle is essential for process optimization, as it allows chemists to fine-tune ligand and oxidant ratios to maximize turnover numbers.
Following the cyclization, the removal of the directing group is achieved through a distinct mechanism involving molybdenum hexacarbonyl. This step is crucial for revealing the final pharmacophore, as the directing group, while essential for the cyclization, is not part of the target bioactive structure. The reaction proceeds in a mixture of acetonitrile and water under a nitrogen atmosphere, conditions that are mild enough to preserve the integrity of the newly formed benzopyran scaffold. The efficiency of this deprotection step is evidenced by the high yields reported in the examples, demonstrating that the guiding group can be cleanly excised without degrading the sensitive alcohol functionality. This dual-catalyst strategy exemplifies a modern approach to complex molecule synthesis, balancing the need for temporary activation with the requirement for clean final products.
How to Synthesize Benzopyran-3-ol Derivatives Efficiently
The synthesis protocol described in the patent offers a clear and reproducible pathway for generating these valuable intermediates, suitable for both laboratory discovery and pilot plant operations. The procedure involves precise control over reaction parameters such as temperature, solvent volume, and catalyst loading to ensure consistent outcomes. Detailed standard operating procedures for mixing, heating, and workup are essential to replicate the high yields observed in the patent examples. For a comprehensive guide on the specific molar ratios and purification techniques required to implement this chemistry, please refer to the standardized synthesis steps outlined below.
- Mix Compound I with a palladium catalyst, oxidant, and solvent, then stir at 70-130°C for 1-24 hours to form Intermediate II.
- Perform post-treatment including filtration and column chromatography to isolate the cyclized intermediate product.
- React Intermediate II with molybdenum hexacarbonyl in acetonitrile and water at 85-95°C to remove the directing group and obtain Product III.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this CDC-based synthesis route offers transformative benefits for procurement strategies and supply chain resilience. By removing the necessity for pre-functionalized starting materials like aryl halides and boronic acids, the process significantly reduces the raw material burden and simplifies the sourcing landscape. This streamlining of the supply chain mitigates risks associated with the availability and price volatility of specialized coupling reagents. Furthermore, the reduction in synthetic steps directly correlates to lower operational expenditures, as fewer unit operations mean reduced labor, energy, and equipment usage. The ability to produce high-purity intermediates with fewer purification challenges also enhances throughput, allowing manufacturers to respond more agilely to market demands.
- Cost Reduction in Manufacturing: The elimination of pre-functionalization steps inherently lowers the cost of goods sold by reducing the consumption of expensive reagents and solvents. Traditional cross-coupling methods often require stoichiometric amounts of organometallic species and generate significant salt waste, whereas this direct C-H activation approach is far more atom-economical. The use of readily available palladium catalysts and common oxidants further contributes to a favorable cost profile compared to proprietary or rare metal systems. Additionally, the simplified workup procedures reduce the time and resources spent on downstream processing, leading to substantial overall cost savings. These economic efficiencies make the process highly attractive for the commercial scale-up of complex pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: Relying on simpler, commodity-grade starting materials enhances the robustness of the supply chain against disruptions. Since the method does not depend on custom-synthesized halides or sensitive organometallics, procurement teams can source inputs from a wider base of suppliers with greater confidence. The mild reaction conditions, operating typically around 90°C, also reduce the energy intensity of the process, making it easier to implement in diverse manufacturing facilities without specialized high-temperature infrastructure. This flexibility ensures a more stable and continuous supply of critical intermediates, safeguarding production schedules against external logistical shocks. Ultimately, a more resilient supply chain translates to better reliability for downstream drug manufacturers.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing solvents and conditions that are manageable on a multi-kilogram to ton scale. The reduction in waste generation aligns with increasingly stringent environmental regulations, lowering the costs associated with waste disposal and treatment. By avoiding the use of highly toxic or hazardous reagents often found in traditional coupling chemistries, the process improves workplace safety and reduces the regulatory burden. The high selectivity of the reaction minimizes the formation of impurities, which simplifies purification and reduces solvent consumption during chromatography or crystallization. These factors collectively support a sustainable manufacturing model that meets both economic and ecological goals.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this synthesis method, derived directly from the patent specifications. These insights are intended to clarify the operational parameters and potential applications for technical teams evaluating this technology. Understanding these details is crucial for assessing the feasibility of integrating this route into existing production workflows. The answers reflect the specific experimental data and conditions reported in the intellectual property documentation.
Q: What are the advantages of this CDC method over traditional Suzuki coupling?
A: This method eliminates the need for pre-functionalized halides and boronic acids, reducing waste and raw material costs while improving atom economy through direct C-H activation.
Q: What are the typical reaction conditions for the cyclization step?
A: The cyclization typically proceeds at mild temperatures between 70-130°C using palladium catalysts and oxidants like N-fluorobisbenzenesulfonamide in solvents such as 1,2-dichloroethane.
Q: How is the directing group removed in the final step?
A: The directing group is efficiently cleaved using molybdenum hexacarbonyl in a mixture of acetonitrile and water under nitrogen protection at 85-95°C.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Benzopyran-3-ol Derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic value of advanced synthetic methodologies like the one described in CN111423405B for accelerating drug development timelines. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative lab-scale chemistry can be successfully translated into robust industrial processes. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of benzopyran-3-ol derivatives meets the exacting standards required by global pharmaceutical clients. Our commitment to technical excellence allows us to navigate the complexities of C-H activation chemistry, delivering high-quality intermediates that support your R&D and commercial manufacturing needs.
We invite you to collaborate with us to leverage this cutting-edge technology for your specific project requirements. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume needs, demonstrating how this efficient route can optimize your budget. Please contact us to request specific COA data and route feasibility assessments, and let us help you secure a reliable supply of these critical building blocks. Together, we can drive innovation and efficiency in the production of next-generation therapeutic agents.