Scalable Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles: A Breakthrough for Commercial API Manufacturing
Scalable Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles: A Breakthrough for Commercial API Manufacturing
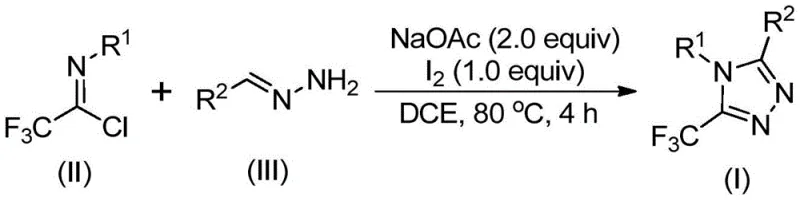
The pharmaceutical and agrochemical industries continuously seek robust, scalable methodologies for constructing nitrogen-containing heterocycles, particularly those incorporating fluorine motifs which enhance metabolic stability and bioavailability. Patent CN110467579B discloses a highly efficient preparation method for 5-trifluoromethyl substituted 1,2,4-triazole compounds, addressing critical bottlenecks in traditional synthetic routes. This technology leverages a non-metallic iodine-promoted cyclization strategy that bypasses the need for harsh anhydrous conditions or expensive transition metal catalysts. The significance of this advancement cannot be overstated, as 1,2,4-triazole scaffolds are ubiquitous in high-value active pharmaceutical ingredients (APIs) ranging from antifungal agents to aromatase inhibitors. By utilizing inexpensive and readily available starting materials such as hydrazones and trifluoroethylimidoyl chloride, this process offers a streamlined pathway for the commercial scale-up of complex pharmaceutical intermediates. The following analysis details the mechanistic advantages and supply chain implications of this novel synthetic approach.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of trifluoromethyl-substituted nitrogen heterocycles has relied heavily on two primary strategies, both of which present significant logistical and economic challenges for large-scale manufacturing. The first approach involves the direct trifluoromethylation of pre-synthesized heterocyclic cores, which necessitates the use of specialized and often hazardous trifluoromethylating reagents that are costly to procure and handle safely on a multi-ton scale. The second mainstream method employs trifluoromethyl synthons, such as trifluorodiazoethane, which pose severe safety risks due to their explosive nature and require stringent safety protocols that inflate operational expenditures. Furthermore, many existing protocols depend on transition metal catalysis, introducing the risk of heavy metal contamination in the final product, a critical failure point for high-purity pharmaceutical intermediate specifications. These conventional routes often demand rigorous anhydrous and anaerobic environments, complicating reactor setup and increasing energy consumption, thereby hindering the cost reduction in API manufacturing that procurement teams desperately seek.
The Novel Approach
In stark contrast, the methodology outlined in patent CN110467579B introduces a paradigm shift by utilizing a metal-free, iodine-promoted cyclization between trifluoroethylimidoyl chloride and hydrazones. This innovative route eliminates the dependency on explosive diazo compounds and expensive metal catalysts, replacing them with benign elemental iodine and simple sodium acetate bases. The reaction operates effectively in common aprotic solvents like dichloroethane (DCE) at moderate temperatures of 80°C to 100°C, removing the necessity for cryogenic cooling or inert atmosphere gloveboxes. This simplicity translates directly into operational resilience, allowing production facilities to utilize standard glass-lined reactors without specialized modifications. The ability to synthesize diversely substituted 1,2,4-triazoles through simple substrate design enhances the versatility of this platform, making it an ideal solution for a reliable pharmaceutical intermediate supplier aiming to offer a broad portfolio of fluorinated building blocks with consistent quality and reduced lead times.
Mechanistic Insights into Iodine-Promoted Cyclization
The mechanistic elegance of this transformation lies in its sequential base-promoted and oxidative steps, which ensure high conversion rates and minimal byproduct formation. The reaction initiates with a base-promoted intermolecular carbon-nitrogen bond formation between the trifluoroethylimidoyl chloride and the hydrazone substrate, generating a trifluoroacetamidine intermediate. This species subsequently undergoes isomerization, setting the stage for the critical oxidative step. The addition of elemental iodine facilitates a base-promoted oxidative iodination, creating a reactive iodine-containing intermediate that is primed for cyclization. This design ensures that the trifluoromethyl group is securely incorporated into the heterocyclic framework early in the sequence, minimizing the risk of defluorination or side reactions that often plague late-stage fluorination strategies. The final stage involves an intramolecular electrophilic substitution followed by aromatization, driving the equilibrium towards the thermodynamically stable 5-trifluoromethyl-1,2,4-triazole product. This clear, stepwise progression allows for precise control over reaction kinetics, ensuring that impurities are kept to a minimum throughout the process.
From an impurity control perspective, the absence of transition metals removes a major class of difficult-to-remove contaminants, simplifying the purification workflow significantly. The use of sodium acetate as a mild base prevents the degradation of sensitive functional groups on the aromatic rings, such as esters or halides, which might be compromised under stronger basic conditions. The oxidative nature of the iodine promotion ensures complete conversion of the intermediate amidines, preventing the accumulation of uncyclized precursors that could complicate downstream isolation. For R&D directors focused on purity profiles, this mechanism offers a distinct advantage by producing a clean crude reaction mixture that requires less aggressive chromatographic separation. The robustness of the catalytic cycle against varying electronic properties of the substrates—whether electron-rich or electron-deficient aryl groups—further underscores the reliability of this chemistry for generating high-purity 1,2,4-triazole derivatives suitable for stringent regulatory filings.
How to Synthesize 5-Trifluoromethyl-1,2,4-Triazole Efficiently
The practical execution of this synthesis is designed for ease of operation, requiring only standard laboratory or plant equipment to achieve high yields. The process begins by combining the key building blocks, trifluoroethylimidoyl chloride and the appropriate hydrazone, with sodium acetate in a solvent like DCE. The mixture is heated to promote the initial condensation, after which elemental iodine is introduced to drive the oxidative cyclization to completion. This straightforward protocol minimizes the number of unit operations, reducing both labor costs and processing time. For detailed operational parameters, stoichiometry, and workup procedures, please refer to the standardized synthesis guide below which outlines the exact steps validated in the patent examples.
- Mix sodium acetate, trifluoroethylimidoyl chloride, and hydrazone in an organic solvent such as dichloroethane (DCE) within a reaction vessel.
- Heat the reaction mixture to 80°C and maintain stirring for 2 to 4 hours to facilitate the initial condensation and cyclization steps.
- Add elemental iodine to the system and continue heating for an additional 1 to 2 hours to promote oxidative aromatization, followed by standard filtration and purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible strategic benefits that extend beyond mere chemical yield. The primary advantage lies in the drastic simplification of the supply chain for raw materials; hydrazones and trifluoroethylimidoyl chlorides are derived from commodity chemicals like aldehydes and amines, which are abundant and price-stable compared to specialized fluorinating reagents. This stability insulates the manufacturing process from the volatility often seen in the market for exotic organofluorine compounds. Furthermore, the elimination of heavy metal catalysts removes the need for expensive scavenging resins or complex extraction protocols designed to meet ppm-level metal specifications, directly contributing to cost reduction in API manufacturing. The operational simplicity also means that production can be scaled rapidly without the need for specialized infrastructure, ensuring reducing lead time for high-purity pharmaceutical intermediates during periods of high demand.
- Cost Reduction in Manufacturing: The economic model of this process is fundamentally superior due to the replacement of costly catalysts and reagents with inexpensive commodity chemicals like iodine and sodium acetate. By avoiding the use of precious metals such as palladium or copper, the process eliminates a significant variable cost component and reduces the burden on waste treatment systems which often charge premiums for heavy metal disposal. Additionally, the reaction does not require energy-intensive anhydrous or anaerobic conditions, allowing for substantial savings in utility costs associated with drying solvents and maintaining inert atmospheres. These cumulative efficiencies result in a lower cost of goods sold (COGS), enabling more competitive pricing strategies for the final active ingredients while maintaining healthy margins.
- Enhanced Supply Chain Reliability: Sourcing reliability is markedly improved because the key starting materials, such as substituted benzaldehydes and hydrazine hydrate, are produced by a wide global network of chemical manufacturers, reducing the risk of single-source bottlenecks. The robustness of the reaction conditions means that production is less susceptible to delays caused by equipment failures related to complex atmospheric controls or sensitive catalyst handling. This resilience ensures a steady flow of material to downstream customers, reinforcing the position of the manufacturer as a reliable pharmaceutical intermediate supplier. The ability to stockpile stable, non-hazardous raw materials further buffers the supply chain against geopolitical or logistical disruptions, guaranteeing continuity of supply for critical medication pipelines.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this method aligns perfectly with modern green chemistry principles by minimizing hazardous waste generation. The absence of toxic heavy metals simplifies the effluent treatment process, reducing the environmental footprint and ensuring compliance with increasingly stringent global regulations regarding metal residues in pharmaceuticals. The scalability is proven by the ability to easily expand the reaction from gram to kilogram scales without losing efficiency, facilitating the commercial scale-up of complex heterocycles. This ease of scale-up allows manufacturers to respond agilely to market demands, transitioning from clinical trial supplies to commercial production volumes with minimal process re-engineering, thus accelerating time-to-market for new drug candidates.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this patented technology. These answers are derived directly from the experimental data and claims within patent CN110467579B, providing clarity on the operational boundaries and capabilities of the synthesis. Understanding these nuances is essential for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios. The responses cover catalyst requirements, reaction conditions, and substrate compatibility to ensure a comprehensive understanding of the process value proposition.
Q: Does this synthesis require expensive heavy metal catalysts?
A: No, the method described in patent CN110467579B utilizes elemental iodine as a promoter instead of toxic or expensive transition metal catalysts, significantly simplifying downstream purification and reducing heavy metal residue risks.
Q: What are the optimal reaction conditions for this triazole formation?
A: The reaction proceeds efficiently in aprotic solvents like dichloroethane (DCE) at temperatures between 80°C and 100°C, without the need for strict anhydrous or anaerobic conditions, making it highly suitable for industrial scale-up.
Q: Can this method accommodate diverse substrate substituents?
A: Yes, the protocol demonstrates excellent functional group tolerance, successfully synthesizing derivatives with various aryl, heteroaryl, and alkenyl substituents at the 4 and 5 positions of the triazole ring.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced heterocyclic intermediates play in the development of next-generation therapeutics. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial reality is seamless. We are committed to delivering stringent purity specifications through our rigorous QC labs, which are equipped to analyze complex fluorinated compounds with precision. By leveraging the efficient, iodine-promoted synthesis described in CN110467579B, we can offer our partners a supply of 5-trifluoromethyl-1,2,4-triazoles that is not only cost-effective but also consistently high in quality, meeting the exacting standards required by global regulatory bodies.
We invite you to engage with our technical procurement team to discuss how this innovative synthetic route can optimize your specific project requirements. Whether you are in the early stages of route scouting or looking to secure a long-term supply for commercial manufacturing, we are prepared to provide a Customized Cost-Saving Analysis tailored to your volume needs. Please contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise in fluorine chemistry can become a cornerstone of your supply chain strategy.