Scalable Synthesis of Third-Generation EGFR Inhibitor Intermediates via Optimized Cyclopropylation
Introduction to Advanced EGFR Inhibitor Synthesis
The development of third-generation Epidermal Growth Factor Receptor (EGFR) tyrosine kinase inhibitors represents a critical frontier in oncology, specifically for treating non-small cell lung cancer (NSCLC) patients who have developed resistance to first-line therapies due to the T790M mutation. Patent CN111606889B discloses a highly efficient preparation method for 4-(1-cyclopropyl-1H-indol-3-yl)-N-phenylpyrimidin-2-amine derivatives, which serve as pivotal intermediates in this therapeutic class. This technology addresses the urgent industry demand for reliable pharmaceutical intermediate supplier capabilities by offering a route that is not only chemically robust but also engineered for industrial viability. The disclosed methodology overcomes significant bottlenecks found in earlier patents, such as WO2016054987, by utilizing commercially accessible starting materials and streamlining purification protocols. For R&D directors and procurement managers, understanding this synthetic pathway is essential for securing a stable supply chain of high-purity API precursors that meet stringent regulatory standards while optimizing production costs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies for synthesizing 1-cyclopropyl-1H-indole derivatives have historically suffered from severe scalability constraints that hinder commercial adoption. For instance, literature methods such as those published in J. Org. Lett. 2008 often rely on excessive amounts of cyclopropylboronic acid, a relatively expensive reagent, which drastically inflates the raw material cost profile. Furthermore, these conventional routes frequently employ toxic and irritating reagents like DMAP (4-Dimethylaminopyridine) and solvents such as toluene, creating substantial environmental, health, and safety (EHS) liabilities for manufacturing facilities. The reaction conditions are also notoriously harsh, often requiring temperatures as high as 95°C, which increases energy consumption and poses thermal safety risks during scale-up. Additionally, the reliance on silica gel column chromatography for purification is a major bottleneck; this technique is feasible for gram-scale laboratory synthesis but is economically and logistically prohibitive for multi-kilogram or ton-scale commercial production due to high solvent usage and waste generation.
The Novel Approach
The innovative process detailed in CN111606889B introduces a paradigm shift by optimizing the cyclopropylation step to be both cost-effective and operationally simple. By fine-tuning the catalyst system to use copper acetate and 2,2'-bipyridine, the reaction proceeds efficiently at a milder temperature of 60°C, significantly reducing energy requirements and thermal stress on the equipment. Crucially, the molar ratio of cyclopropylboronic acid is optimized to between 1:1 and 1:1.5 relative to the substrate, minimizing reagent waste and cost without sacrificing conversion efficiency. 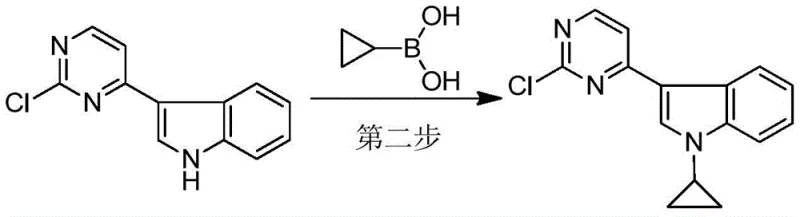 The most transformative aspect of this novel approach is the elimination of silica gel purification; instead, the process utilizes a straightforward recrystallization protocol using ethanol, which dramatically shortens the operation cycle and reduces the volume of hazardous waste solids and liquids. This shift from chromatography to crystallization is a key enabler for cost reduction in pharmaceutical intermediate manufacturing, allowing for seamless transition from pilot plant to full commercial scale.
The most transformative aspect of this novel approach is the elimination of silica gel purification; instead, the process utilizes a straightforward recrystallization protocol using ethanol, which dramatically shortens the operation cycle and reduces the volume of hazardous waste solids and liquids. This shift from chromatography to crystallization is a key enabler for cost reduction in pharmaceutical intermediate manufacturing, allowing for seamless transition from pilot plant to full commercial scale.
Mechanistic Insights into Copper-Catalyzed Cyclopropylation
The core chemical transformation in this synthesis is the copper-catalyzed coupling of the indole-pyrimidine scaffold with cyclopropylboronic acid, a variation of the Chan-Lam coupling reaction. Mechanistically, this involves the activation of the boronic acid by the copper catalyst in the presence of a base such as sodium carbonate or potassium phosphate. The reaction is conducted in polar aprotic solvents like acetonitrile or tetrahydrofuran, which facilitate the solubility of the inorganic base and the organic substrates. The choice of ligand, specifically 2,2'-bipyridine, is critical for stabilizing the copper species and promoting the transmetallation step where the cyclopropyl group is transferred to the copper center. This is followed by coordination with the nitrogen atom of the indole ring and subsequent reductive elimination to form the N-cyclopropyl bond. The patent specifies that maintaining the reaction temperature between 50°C and 65°C is optimal; temperatures below this range may lead to incomplete conversion, while higher temperatures could promote decomposition of the sensitive boronic acid or side reactions. The precise control of stoichiometry, with a preferred molar ratio of substrate to catalyst of 1:1, ensures that the catalytic cycle turns over efficiently without requiring large excesses of metal, which simplifies downstream metal removal processes.
Impurity control is another vital aspect of the mechanistic design, particularly given the sensitivity of the acrylamide warhead installed in later steps. The initial Grignard reaction used to attach the pyrimidine ring to the indole core is performed at 0°C initially to control exothermicity, then heated to 70°C to drive completion. This two-stage temperature profile minimizes the formation of regioisomers or over-alkylated byproducts. Subsequent purification via recrystallization from ethyl acetate and heptane effectively removes unreacted starting materials and inorganic salts. The final acrylamide formation involves an elimination reaction under basic conditions, where careful pH control during workup prevents polymerization of the acrylamide double bond. 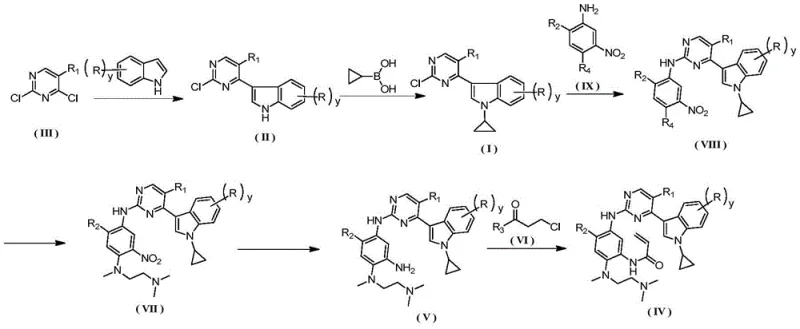 The entire sequence is designed to maintain high chemical integrity, with HPLC purity consistently exceeding 99% for key intermediates, ensuring that the final API meets the rigorous impurity specifications required for oncology drugs.
The entire sequence is designed to maintain high chemical integrity, with HPLC purity consistently exceeding 99% for key intermediates, ensuring that the final API meets the rigorous impurity specifications required for oncology drugs.
How to Synthesize 4-(1-cyclopropyl-1H-indol-3-yl) Derivatives Efficiently
The synthesis of these complex heterocyclic compounds requires a disciplined approach to reaction engineering and process control to ensure reproducibility and safety. The patented route is divided into distinct operational units, beginning with the construction of the chloro-pyrimidine-indole core, followed by N-alkylation, aromatic nucleophilic substitution, reduction, and finally acylation. Each step has been optimized to maximize yield and minimize the accumulation of impurities that could be difficult to remove in later stages. For example, the reduction of the nitro group to the amine is performed using Raney Nickel under hydrogen pressure, a cost-effective alternative to precious metal catalysts like Palladium on Carbon, which aligns with the goal of cost reduction in pharmaceutical intermediate manufacturing. The final salt formation with methanesulfonic acid is conducted in a mixed solvent system of acetone and water, which promotes the precipitation of the product in a highly crystalline form, facilitating easy filtration and drying. Detailed standardized synthesis steps for implementing this route are provided in the guide below.
- React indole with 2,4-dichloropyrimidine using methylmagnesium bromide in THF to form 3-(2-chloropyrimidin-4-yl)-1H-indole.
- Perform copper-catalyzed cyclopropylation with cyclopropylboronic acid at 60°C to yield 3-(2-chloropyrimidin-4-yl)-1-cyclopropyl-1H-indole.
- Couple the chloro-intermediate with a substituted nitro-aniline in 2-pentanol with TsOH catalyst to form the pyrimidin-2-amine scaffold.
- Substitute the fluoro-group with N,N,N'-trimethylethylenediamine to introduce the side chain, followed by catalytic hydrogenation to reduce the nitro group.
- React the resulting triamine with 3-chloropropionyl chloride followed by elimination to install the acrylamide warhead.
- Purify the final product via recrystallization and convert to the methanesulfonate salt using methanesulfonic acid in acetone/water.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the technical improvements in this patent translate directly into tangible commercial benefits that enhance supply security and margin potential. The elimination of silica gel chromatography is perhaps the most significant advantage, as it removes a major bottleneck that typically limits batch size and extends production lead times. By switching to recrystallization, the process becomes amenable to continuous processing or large batch reactors, significantly increasing throughput capacity without proportional increases in labor or equipment costs. Furthermore, the avoidance of toxic solvents like toluene and reagents like DMAP simplifies waste disposal protocols and reduces the regulatory burden associated with handling hazardous materials, thereby lowering the total cost of ownership for the manufacturing site. These factors collectively contribute to a more resilient supply chain capable of meeting the fluctuating demands of the global oncology market.
- Cost Reduction in Manufacturing: The optimized stoichiometry of cyclopropylboronic acid reduces raw material expenses significantly compared to literature methods that require large excesses. Additionally, the replacement of expensive palladium catalysts with copper systems for the coupling step and Raney Nickel for reduction lowers catalyst costs. The simplified workup procedures, which avoid column chromatography, drastically reduce solvent consumption and waste treatment costs, leading to substantial overall cost savings in the production of these high-value intermediates.
- Enhanced Supply Chain Reliability: The reliance on commercially available and stable starting materials, such as indole and 2,4-dichloropyrimidine, mitigates the risk of raw material shortages that can plague specialized synthetic routes. The robustness of the reaction conditions, which do not require strict moisture control or cryogenic temperatures for the coupling steps, ensures consistent batch-to-batch quality and reduces the likelihood of production failures. This reliability is crucial for maintaining uninterrupted supply to downstream API manufacturers and ensuring timely delivery to pharmaceutical clients.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, with reaction temperatures and pressures that are easily managed in standard glass-lined or stainless steel reactors. The reduction in hazardous waste generation through the elimination of silica gel and toxic solvents aligns with modern green chemistry principles and increasingly strict environmental regulations. This compliance not only avoids potential fines but also enhances the corporate sustainability profile, making the supply chain more attractive to environmentally conscious partners and investors.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and advantageous effects reported in the patent documentation, providing a clear understanding of the process capabilities. Understanding these details is vital for technical teams evaluating the feasibility of adopting this route for their own production needs or for sourcing these intermediates from external partners. The clarity on purity, safety, and scalability helps in making informed decisions regarding technology transfer and vendor qualification.
Q: How does this synthesis method improve scalability compared to prior art?
A: The patented method eliminates the need for silica gel column chromatography, replacing it with efficient recrystallization steps. It also operates at milder temperatures (60°C vs 95°C) and avoids toxic solvents like toluene and reagents like DMAP, significantly reducing environmental pressure and operational complexity for large-scale manufacturing.
Q: What purity levels can be achieved with this process?
A: The process consistently delivers intermediates and final products with HPLC purity exceeding 99%. For instance, the key cyclopropylated intermediate achieves 99.9% purity after recrystallization, ensuring a clean impurity profile critical for pharmaceutical regulatory compliance.
Q: Is the cyclopropylation step cost-effective for industrial production?
A: Yes, the method optimizes the molar ratio of cyclopropylboronic acid to 1:1~1.5, significantly reducing reagent costs compared to literature methods requiring large excesses. The use of readily available copper acetate and bipyridine catalysts further enhances cost efficiency without compromising yield.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4-(1-cyclopropyl-1H-indol-3-yl)-N-phenylpyrimidin-2-amine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality intermediates in the development of life-saving oncology therapies. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to market supply is seamless and efficient. We adhere to stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instrumentation to guarantee that every batch of 4-(1-cyclopropyl-1H-indol-3-yl)-N-phenylpyrimidin-2-amine derivatives meets the highest industry standards. Our commitment to quality and consistency makes us a trusted partner for pharmaceutical companies seeking to secure their supply chain for third-generation EGFR inhibitors.
We invite you to engage with our technical procurement team to discuss how this optimized synthesis route can benefit your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic advantages of this method compared to your current supply sources. We encourage potential partners to contact us for specific COA data and route feasibility assessments, allowing us to demonstrate our capability to deliver high-purity intermediates that accelerate your drug development timelines while optimizing your overall production costs.