Scalable Manufacturing of Third-Generation EGFR Inhibitor Intermediates via Optimized Coupling
Introduction to Advanced EGFR Inhibitor Intermediate Manufacturing
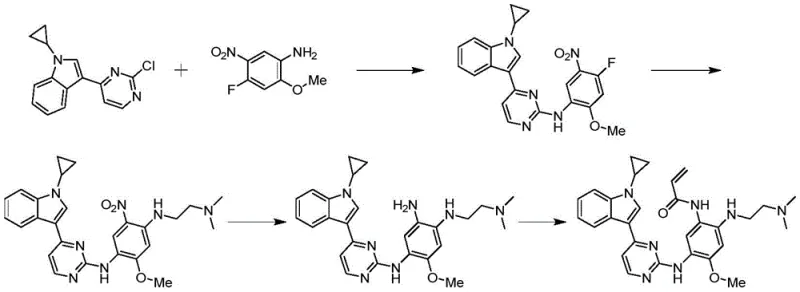
The development of third-generation Epidermal Growth Factor Receptor (EGFR) tyrosine kinase inhibitors represents a pivotal advancement in oncology therapeutics, specifically targeting resistant mutations such as T790M in non-small cell lung cancer (NSCLC). Patent CN111606889A discloses a highly efficient preparation method for 4-(1-cyclopropyl-1H-indol-3-yl)-N-phenylpyrimidin-2-amine derivatives, which serve as critical scaffolds for these next-generation drugs. Unlike traditional pathways that rely on scarce precursors or hazardous conditions, this innovation introduces a robust synthetic strategy that prioritizes operational safety and product integrity. By leveraging a novel sequence of Grignard activation followed by copper-catalyzed cyclopropylation, the process addresses the longstanding supply chain bottlenecks associated with complex heterocyclic intermediates. This technical breakthrough not only enhances the chemical purity required for stringent regulatory filings but also establishes a foundation for reliable pharmaceutical intermediate supplier partnerships focused on long-term commercial viability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies, such as those described in WO2016054987, often depended on 3-(2-chloropyrimidin-4-yl)-1-cyclopropyl-1H-indole as a starting material, a compound that is notoriously difficult to procure in bulk quantities suitable for industrial campaigns. Furthermore, academic precedents like those in J. Org. Lett. 2008 utilized cyclopropylboronic acid in large excess, driving up raw material costs significantly due to the reagent's high market price. These legacy processes frequently employed toxic catalysts such as DMAP (4-Dimethylaminopyridine) and irritant solvents like toluene, creating substantial environmental compliance burdens and worker safety risks. Additionally, reaction conditions often required elevated temperatures around 95°C, increasing energy consumption and the potential for thermal degradation of sensitive intermediates. The reliance on silica gel column chromatography for purification in earlier methods further exacerbated waste generation, producing large volumes of solid and liquid waste that complicate disposal and increase the overall cost reduction in API manufacturing efforts.
The Novel Approach
The innovative pathway outlined in CN111606889A fundamentally restructures the synthesis to overcome these inefficiencies by starting from readily available indole and 2,4-dichloropyrimidine. This strategy employs a Grignard reagent to activate the indole nucleus, facilitating a smooth coupling with the pyrimidine ring under controlled thermal conditions. A key differentiator is the optimized copper-catalyzed cyclopropylation step, which operates at a milder temperature range of 50°C to 65°C, significantly lowering energy inputs while maintaining high conversion rates. Crucially, the process replaces labor-intensive silica gel purification with a streamlined recrystallization protocol using ethanol, which drastically shortens the operation cycle and minimizes waste solids. This approach ensures that the commercial scale-up of complex pharmaceutical intermediates is not only chemically feasible but also economically sustainable, providing a clear advantage for procurement teams seeking stable supply chains.
Mechanistic Insights into Copper-Catalyzed N-Arylation and Grignard Activation
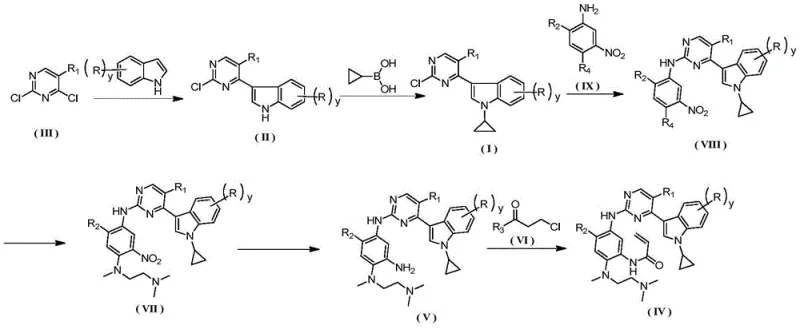
The core of this synthetic success lies in the precise orchestration of organometallic transformations, beginning with the regioselective activation of the indole ring. The initial step utilizes methylmagnesium bromide to deprotonate the indole nitrogen, generating a nucleophilic species that attacks the 4-position of 2,4-dichloropyrimidine. This Grignard-mediated addition is critical for establishing the core indole-pyrimidine linkage with high fidelity, avoiding the formation of unwanted regioisomers that could compromise downstream purity. Following this, the introduction of the cyclopropyl group is achieved through a Chan-Lam-type coupling mechanism involving cyclopropylboronic acid and a copper catalyst system. The use of copper acetate combined with 2,2'-bipyridine ligands facilitates the oxidative addition and reductive elimination cycles necessary for C-N bond formation without requiring harsh alkylating agents. This catalytic cycle is remarkably tolerant of functional groups, allowing for the preservation of the chloro-pyrimidine moiety which is essential for subsequent nucleophilic aromatic substitution reactions with aniline derivatives.
Impurity control is rigorously managed through the thermodynamic properties of the intermediates, particularly during the isolation of 3-(2-chloropyrimidin-4-yl)-1-cyclopropyl-1H-indole. By optimizing the solvent system to include ethanol for recrystallization, the process leverages solubility differences to exclude trace metal residues and unreacted boronic acid species. The reduction of the nitro group in later stages utilizes Raney-Ni or Pd/C under hydrogen pressure, a standard yet highly effective method for converting nitro-anilines to diamines without affecting the sensitive acrylamide precursor functionality. The final acryloylation step is conducted at low temperatures (0°C to 10°C) to prevent polymerization of the acrylic double bond, ensuring the structural integrity of the pharmacophore. This meticulous attention to reaction kinetics and thermodynamics throughout the seven-step sequence guarantees that the final active pharmaceutical ingredient precursors meet the stringent purity specifications demanded by global regulatory bodies.
How to Synthesize 4-(1-Cyclopropyl-1H-indol-3-yl) Derivatives Efficiently
Executing this synthesis requires strict adherence to the optimized parameters defined in the patent to ensure reproducibility and safety at scale. The process begins with the preparation of the chloro-pyrimidine indole scaffold, followed by the critical cyclopropylation which sets the stereochemical and electronic properties of the molecule. Subsequent steps involve sequential nucleophilic substitutions and reductions that build the complex side chains necessary for EGFR inhibition. Operators must maintain inert atmospheres during Grignard additions and carefully control exotherms during the acryloylation phase to prevent runaway reactions. The detailed standardized synthesis steps see the guide below for specific molar ratios and temperature profiles that have been validated for industrial throughput.
- React indole with methylmagnesium bromide and 2,4-dichloropyrimidine in THF at 70°C to form 3-(2-chloropyrimidin-4-yl)-1H-indole.
- Perform copper-catalyzed coupling with cyclopropylboronic acid at 60°C using sodium carbonate and bipyridine to yield 3-(2-chloropyrimidin-4-yl)-1-cyclopropyl-1H-indole.
- Condense with nitro-aniline derivatives in 2-pentanol with TsOH catalyst at 110°C, followed by diamine substitution, nitro reduction, and final acryloylation.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this novel manufacturing process offers tangible strategic benefits beyond mere chemical elegance. The primary advantage is the substantial cost savings derived from the elimination of expensive and toxic reagents like DMAP and the reduction of solvent usage through efficient recrystallization techniques. By shifting away from silica gel chromatography, the facility significantly lowers its hazardous waste disposal costs and reduces the turnaround time between batches, thereby enhancing overall equipment effectiveness. This efficiency translates directly into a more competitive pricing structure for the final intermediate, allowing pharmaceutical partners to optimize their own cost reduction in API manufacturing budgets without compromising on quality standards.
- Cost Reduction in Manufacturing: The process achieves significant economic optimization by utilizing commercially abundant starting materials such as indole and dichloropyrimidine, which are far less expensive than the specialized pre-functionalized intermediates required by older methods. The removal of the silica gel purification step eliminates the cost of stationary phases and the associated solvent volumes needed for elution, leading to a drastic reduction in raw material expenditure. Furthermore, the milder reaction temperatures reduce utility costs associated with heating and cooling large-scale reactors, contributing to a leaner operational budget. These cumulative efficiencies ensure that the production of high-purity pharmaceutical intermediates remains financially viable even under fluctuating market conditions for fine chemicals.
- Enhanced Supply Chain Reliability: Sourcing stability is greatly improved because the synthesis relies on commodity chemicals that are widely available from multiple global vendors, mitigating the risk of single-source bottlenecks. The robustness of the copper-catalyzed coupling step means that minor variations in raw material quality do not lead to batch failures, ensuring consistent output volumes for downstream drug substance manufacturing. This reliability is crucial for maintaining continuous supply lines for life-saving oncology medications, where interruptions can have severe clinical consequences. By adopting this method, partners secure a resilient supply chain capable of withstanding logistical disruptions and meeting rigorous delivery schedules.
- Scalability and Environmental Compliance: The design of this synthetic route inherently supports commercial scale-up of complex pharmaceutical intermediates from pilot plants to multi-ton production facilities. The replacement of toxic solvents like toluene with greener alternatives such as 2-pentanol and ethanol aligns with increasingly strict environmental regulations and corporate sustainability goals. The reduction in solid waste generation through crystallization-based purification simplifies waste treatment protocols and lowers the environmental footprint of the manufacturing site. This commitment to green chemistry principles not only ensures regulatory compliance but also enhances the brand reputation of the supply chain partners involved in the production of these vital therapeutic agents.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. They are derived from the specific advantages and operational details outlined in the patent documentation to provide clarity for potential partners. Understanding these aspects is essential for evaluating the feasibility of integrating this intermediate into your existing drug development pipeline.
Q: How does this process improve upon previous synthesis methods for EGFR intermediates?
A: This method eliminates the need for difficult-to-procure starting materials and replaces toxic reagents like DMAP and solvents like toluene with safer alternatives. It also substitutes silica gel purification with recrystallization, significantly reducing solid waste and operational time.
Q: What are the critical purity specifications achieved in this synthesis?
A: The optimized process consistently achieves HPLC purity levels exceeding 99% for key intermediates and the final product. For instance, the cyclopropylated indole intermediate reaches 99.9% purity through simple ethanol recrystallization.
Q: Is this synthetic route suitable for large-scale commercial production?
A: Yes, the route is designed for industrial scalability. It utilizes mild reaction temperatures (50-60°C for coupling), avoids moisture-sensitive steps where possible, and employs robust workup procedures like filtration and crystallization that are easily transferable to multi-ton reactors.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4-(1-Cyclopropyl-1H-indol-3-yl)-N-phenylpyrimidin-2-amine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the successful development of third-generation EGFR inhibitors. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition smoothly from clinical trials to market launch. We are committed to delivering products with stringent purity specifications and rigorous QC labs testing every batch to guarantee consistency and safety. Our state-of-the-art facilities are equipped to handle the specific reaction conditions required for copper-catalyzed couplings and sensitive acryloylations, providing a secure environment for your proprietary chemistry.
We invite you to contact our technical procurement team to discuss how we can support your specific volume requirements and timeline goals. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how our optimized process can reduce your overall COGS. We are ready to provide specific COA data and route feasibility assessments to demonstrate our capability as a trusted partner in your supply chain. Let us collaborate to bring these life-saving therapies to patients faster and more efficiently.