Advanced Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles for Scalable Pharmaceutical Manufacturing
Advanced Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles for Scalable Pharmaceutical Manufacturing
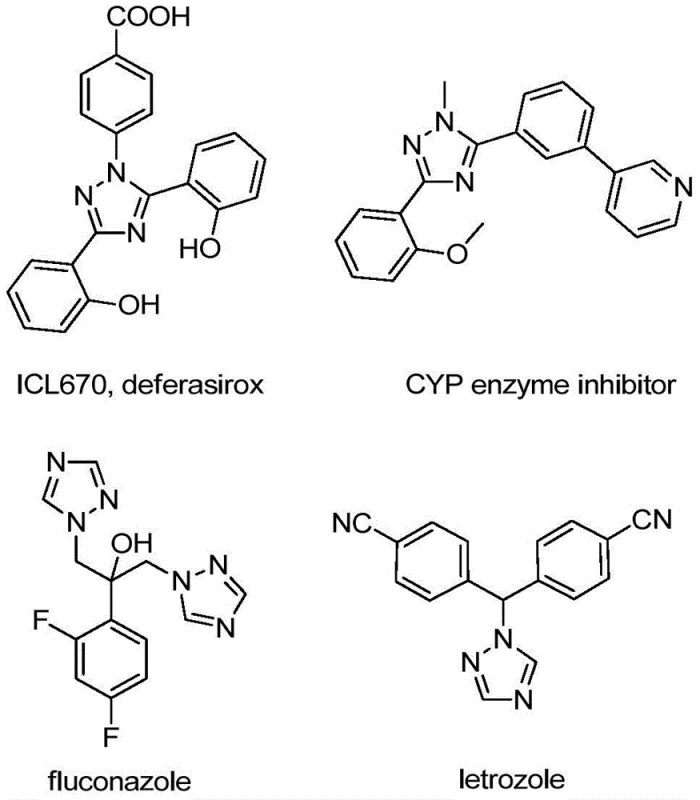
The integration of trifluoromethyl groups into heterocyclic scaffolds represents a cornerstone strategy in modern medicinal chemistry, profoundly enhancing the metabolic stability, lipophilicity, and bioavailability of drug candidates. As detailed in the groundbreaking patent CN110467579B, a novel and highly efficient preparation method for 5-trifluoromethyl substituted 1,2,4-triazole compounds has been developed, addressing critical bottlenecks in the supply chain of high-value pharmaceutical intermediates. This technology leverages a non-metallic iodine-promoted cyclization strategy that bypasses the limitations of traditional trifluoromethylation reagents, offering a streamlined pathway for the commercial scale-up of complex pharmaceutical intermediates. The significance of this innovation cannot be overstated for R&D teams seeking reliable routes to fluorinated heterocycles, which are ubiquitous in blockbuster drugs ranging from antifungals to kinase inhibitors.
The structural versatility of the 1,2,4-triazole ring makes it a privileged scaffold in drug design, yet introducing the trifluoromethyl moiety at the 5-position has historically been fraught with synthetic challenges. The methodology disclosed in this patent utilizes inexpensive and readily available hydrazones and trifluoroethylimidoyl chlorides as starting materials, reacting them under mild thermal conditions to yield the desired products with high efficiency. This approach not only simplifies the synthetic route but also drastically reduces the environmental footprint by eliminating the need for hazardous reagents often associated with fluorination chemistry. For procurement managers and supply chain heads, this translates to a more resilient sourcing strategy for critical building blocks used in the synthesis of active pharmaceutical ingredients (APIs).
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of trifluoromethyl-substituted nitrogen-containing heterocycles has relied heavily on two primary strategies, both of which present significant drawbacks for large-scale manufacturing. The first conventional approach involves the direct trifluoromethylation of pre-synthesized nitrogen heterocycles, a process that typically necessitates the use of specialized and often expensive trifluoromethylating reagents. These reagents can be unstable, difficult to handle, and generate substantial amounts of hazardous waste, complicating the post-reaction purification processes and driving up the overall cost of goods. Furthermore, the second mainstream method utilizes synthons bearing trifluoromethyl groups, such as trifluorodiazoethane, which reacts with coupling substrates. While effective in academic settings, trifluorodiazoethane is notoriously explosive and requires stringent safety protocols, making it ill-suited for multi-kilogram or ton-scale production in standard chemical plants.
Another common synthon, trifluoroethylimide acid halide, has seen relatively limited application due to perceived difficulties in controlling regioselectivity and reaction efficiency. Traditional methods often demand rigorous anhydrous and anaerobic conditions, requiring specialized equipment like gloveboxes or Schlenk lines, which adds considerable capital expenditure and operational complexity. Additionally, many existing protocols rely on transition metal catalysts, which introduce the risk of heavy metal contamination in the final product. For pharmaceutical manufacturers, removing trace metals to meet stringent regulatory limits (often in the parts per million range) requires additional purification steps, such as scavenging or recrystallization, further eroding profit margins and extending lead times for high-purity pharmaceutical intermediates.
The Novel Approach
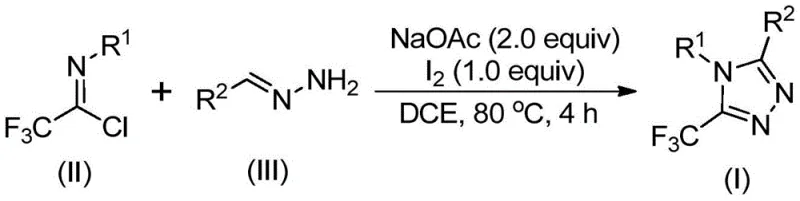
In stark contrast to these legacy methods, the technology described in patent CN110467579B introduces a paradigm shift by employing a simple, metal-free cyclization strategy promoted by elemental iodine. This novel approach utilizes trifluoroethylimidoyl chloride and hydrazones as the core building blocks, reacting them in the presence of sodium acetate in an organic solvent. The reaction proceeds smoothly at moderate temperatures, typically around 80°C, without the need for inert atmosphere protection or strictly anhydrous conditions. This robustness is a game-changer for industrial applicability, as it allows the reaction to be performed in standard stainless steel reactors without the need for exotic lining or complex gas handling systems. The use of elemental iodine as a promoter is particularly advantageous; it is inexpensive, readily available, and easy to remove during workup, unlike precious metal catalysts.
Moreover, this method exhibits exceptional substrate tolerance, allowing for the synthesis of a wide variety of 5-trifluoromethyl-1,2,4-triazole derivatives with different substituents at the 4 and 5 positions. By simply varying the R1 and R2 groups on the starting hydrazone and imidoyl chloride, chemists can access a diverse library of compounds tailored for specific biological targets. The operational simplicity extends to the workup procedure, which involves basic filtration and column chromatography, avoiding complex extraction sequences. This streamlined workflow significantly reduces the time from bench to batch, enabling faster iteration cycles for R&D teams and quicker time-to-market for new drug candidates. The ability to scale this reaction to the gram level and beyond, as demonstrated in the patent examples, underscores its potential for seamless technology transfer to commercial manufacturing facilities.
Mechanistic Insights into Iodine-Promoted Cyclization
The mechanistic pathway of this iodine-promoted synthesis offers fascinating insights into how mild conditions can achieve high transformation efficiency. The reaction is believed to initiate with a base-promoted intermolecular carbon-nitrogen bond formation between the trifluoroethylimidoyl chloride and the hydrazone, generating a trifluoroacetamidine intermediate. This initial condensation is facilitated by sodium acetate, which acts as a mild base to scavenge the hydrochloric acid byproduct, driving the equilibrium forward. Following this, the intermediate undergoes isomerization, setting the stage for the critical cyclization step. The addition of elemental iodine then triggers a base-promoted oxidative iodination, forming an iodinated intermediate that is highly reactive towards intramolecular nucleophilic attack.
This intramolecular electrophilic substitution is the key ring-closing event, where the nitrogen atom attacks the activated carbon center, closing the five-membered triazole ring. The final step involves aromatization, likely driven by the elimination of hydrogen iodide, to yield the stable 5-trifluoromethyl substituted 1,2,4-triazole product. Understanding this mechanism is crucial for process optimization, as it highlights the dual role of the base in both the initial condensation and the oxidative cyclization. From an impurity control perspective, the mild nature of the reagents minimizes the formation of side products such as over-oxidized species or polymerization byproducts that are common in harsher radical trifluoromethylation reactions. The specificity of the iodine promotion ensures that the reaction proceeds through a defined ionic pathway rather than a chaotic radical chain, resulting in cleaner reaction profiles and higher crude purity.
Furthermore, the electronic properties of the trifluoromethyl group play a significant role in stabilizing the intermediate species and directing the regioselectivity of the cyclization. The strong electron-withdrawing nature of the CF3 group enhances the electrophilicity of the imidoyl carbon, facilitating the initial nucleophilic attack by the hydrazone. This electronic activation allows the reaction to proceed at lower temperatures compared to non-fluorinated analogs, reducing energy consumption and thermal stress on the equipment. For quality control teams, the predictability of this mechanism means that impurity profiles are consistent and manageable, simplifying the validation of analytical methods for release testing. The ability to fine-tune the electronic environment by modifying the aryl substituents on the starting materials provides an additional layer of control over reaction kinetics and product distribution.
How to Synthesize 5-Trifluoromethyl-1,2,4-Triazole Efficiently
The practical execution of this synthesis is designed for ease of operation, making it accessible even to laboratories with standard equipment. The protocol begins with the combination of sodium acetate, trifluoroethylimidoyl chloride, and the appropriate hydrazone in a suitable organic solvent such as 1,2-dichloroethane (DCE). The mixture is heated to approximately 80°C and stirred for 2 to 4 hours to ensure complete conversion to the amidine intermediate. Subsequently, elemental iodine is added directly to the reaction vessel, and heating is continued for an additional 1 to 2 hours to drive the cyclization to completion. The detailed standardized synthesis steps, including precise molar ratios and workup procedures, are outlined in the guide below.
- Mix sodium acetate, trifluoroethylimidoyl chloride, and hydrazone in an organic solvent such as dichloroethane.
- Heat the reaction mixture to 80°C and stir for 2 to 4 hours to allow initial condensation.
- Add elemental iodine to the system and continue heating for an additional 1 to 2 hours to promote cyclization.
- Filter the mixture, mix with silica gel, and purify via column chromatography to obtain the final triazole product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthesis method offers tangible strategic advantages that extend beyond mere chemical yield. The primary value proposition lies in the drastic simplification of the supply chain for raw materials. Unlike methods requiring specialized fluorinating agents or unstable diazo compounds, this process relies on trifluoroethylimidoyl chlorides and hydrazones, which are commodity chemicals available from multiple global suppliers. This diversification of the supplier base mitigates the risk of single-source dependency and price volatility, ensuring a steady flow of materials for continuous manufacturing operations. The elimination of heavy metal catalysts further simplifies the procurement landscape, removing the need for sourcing expensive palladium or copper salts and the associated ligands.
- Cost Reduction in Manufacturing: The economic impact of this technology is profound, primarily driven by the removal of costly purification steps associated with heavy metal removal. In traditional catalytic processes, significant resources are allocated to scavenging resins, activated carbon treatments, or extensive recrystallizations to meet residual metal specifications. By utilizing an iodine-promoted, metal-free system, these downstream processing costs are virtually eliminated, leading to substantial cost savings in the overall manufacturing budget. Additionally, the use of inexpensive elemental iodine and sodium acetate as reagents keeps the direct material costs low, while the high atom economy of the cyclization reaction minimizes waste disposal fees. The ability to run the reaction without strict anhydrous conditions also reduces utility costs related to solvent drying and nitrogen purging.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions directly translates to improved supply chain reliability and reduced lead times. Since the process does not require sensitive handling or specialized infrastructure, it can be manufactured in a wider range of facilities, increasing the available capacity in the market. The stability of the starting materials allows for longer storage times and easier transportation, reducing the risk of degradation during logistics. This reliability is critical for maintaining just-in-time inventory levels for API production, preventing costly shutdowns due to intermediate shortages. Furthermore, the scalability of the method ensures that supply can be rapidly ramped up to meet surges in demand without the need for lengthy process re-validation.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this method aligns perfectly with modern green chemistry principles and regulatory expectations. The avoidance of explosive reagents like trifluorodiazoethane significantly lowers the safety risk profile of the manufacturing site, potentially reducing insurance premiums and safety compliance costs. The simpler waste stream, devoid of heavy metals, facilitates easier treatment and disposal, helping companies meet increasingly stringent environmental regulations. The process is inherently scalable, as demonstrated by its successful expansion to gram scales in the patent, suggesting a smooth path to kilogram and ton-scale production. This scalability ensures that the technology remains viable as the drug candidate progresses from clinical trials to commercial launch.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this 5-trifluoromethyl-1,2,4-triazole synthesis technology. These answers are derived directly from the experimental data and technical specifications provided in the patent documentation, ensuring accuracy and relevance for decision-makers evaluating this route for their pipeline.
Q: Does this synthesis require expensive heavy metal catalysts?
A: No, the method described in patent CN110467579B utilizes elemental iodine as a promoter, completely avoiding the need for costly and toxic transition metal catalysts like palladium or copper.
Q: What are the reaction conditions regarding moisture and oxygen?
A: The process is robust and does not require strict anhydrous or anaerobic conditions, significantly simplifying operational requirements compared to traditional trifluoromethylation methods.
Q: Is this method suitable for large-scale production?
A: Yes, the patent explicitly states that the method can be easily expanded to the gram level and beyond, utilizing cheap and readily available starting materials suitable for industrial application.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the success of your drug development programs. Our team of expert process chemists has extensively evaluated the technology described in patent CN110467579B and possesses the capability to implement this efficient, iodine-promoted synthesis at commercial scale. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with consistency and precision. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 5-trifluoromethyl-1,2,4-triazole delivered meets the highest industry standards for pharmaceutical applications.
We invite you to collaborate with us to leverage this advanced synthetic route for your next project. By partnering with NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact our technical procurement team today to request specific COA data for our reference standards and to discuss route feasibility assessments for your custom synthesis needs. Let us help you optimize your supply chain and accelerate your path to market with our reliable and cost-effective manufacturing solutions.