Advanced Synthesis of 3,4-Dihydro-2H-quinolizin-2-one Derivatives for Pharmaceutical Applications
The pharmaceutical and agrochemical industries are constantly seeking novel nitrogen-containing heterocyclic scaffolds that offer unique biological activities and improved pharmacokinetic profiles. A recent breakthrough detailed in patent CN114478516B introduces a sophisticated class of 3,4-dihydro-2H-quinolizin-2-one compounds, representing a significant advancement in organic synthesis methodology. These compounds are not merely theoretical constructs but serve as vital building blocks for potential drug candidates and pesticide formulations, addressing a long-standing gap in the availability of diverse quinolizinone derivatives. The patent discloses a robust preparation method that transforms readily available 2-pyridine acetate derivatives into these complex bicyclic systems through an elegant intramolecular electrocyclization dearomatization reaction. This innovation is particularly noteworthy for its ability to generate structural diversity through simple substituent variations on the aryl and pyridine rings, providing medicinal chemists with a powerful toolkit for lead optimization. As a reliable pharmaceutical intermediate supplier, understanding the nuances of such novel scaffolds is crucial for developing next-generation therapeutics.
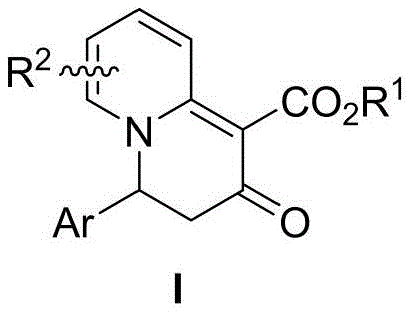
The significance of this technology extends beyond mere structural novelty; it offers a streamlined pathway to access chemical space that was previously difficult to navigate. Nitrogen heterocycles are ubiquitous in bioactive molecules, with statistics showing that a vast majority of FDA-approved small molecule drugs contain at least one such ring system. However, the specific 3,4-dihydro-2H-quinolizin-2-one core has remained elusive due to synthetic challenges associated with maintaining the integrity of the fused ring system while introducing functional handles. The disclosed method overcomes these hurdles by leveraging the unique reactivity of (2Z,4E)-2-(2-pyridyl)-3-hydroxy-5-aryl-2,4-dienoate precursors. By utilizing mild reaction conditions and avoiding harsh reagents, this process ensures high purity and minimizes the formation of complex impurity profiles, which is a critical consideration for regulatory compliance in API manufacturing. This approach exemplifies the kind of process innovation that drives cost reduction in pharmaceutical intermediate manufacturing by simplifying the synthetic route.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of quinolizinone frameworks has been plagued by significant synthetic inefficiencies and operational complexities. Conventional routes often rely on multi-step sequences involving expensive transition metal catalysts, stringent anhydrous conditions, or high-temperature cyclizations that can degrade sensitive functional groups. Literature surveys indicate that while methods exist for related indolizine or 2,3-dihydro-4H-quinazin-4-one systems, the specific synthesis of the 3,4-dihydro-2H-quinolizin-2-one motif has been largely unreported or achieved with poor yields. Traditional dearomatization strategies frequently require stoichiometric amounts of oxidants or activators, generating substantial chemical waste and complicating downstream purification. Furthermore, the use of heavy metal catalysts introduces the risk of residual metal contamination, necessitating costly and time-consuming scavenging steps to meet strict pharmaceutical specifications. These limitations create bottlenecks in the supply chain, increasing lead times and reducing the overall economic viability of producing these valuable intermediates for drug discovery programs.
The Novel Approach
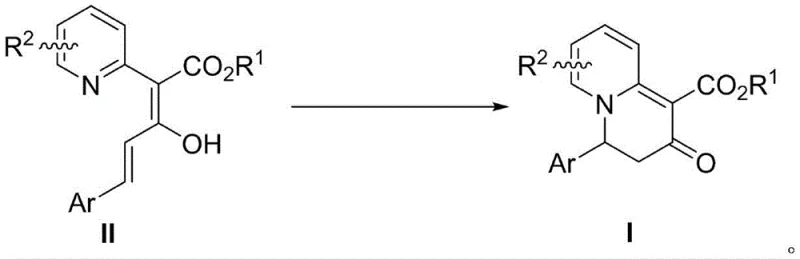
In stark contrast to these cumbersome traditional methods, the novel approach described in the patent utilizes a catalyst-free or mild acid-catalyzed intramolecular electrocyclization that proceeds under remarkably benign conditions. The core transformation involves dissolving the 2-pyridine acetate derivative precursor in a specialized solvent system, specifically hexafluoroisopropanol, and allowing the reaction to stir at room temperature. This eliminates the need for energy-intensive heating or cooling cycles, drastically simplifying the operational requirements for commercial scale-up. The reaction mechanism leverages the inherent electronic properties of the conjugated dienoate system to trigger a spontaneous ring closure, effectively converting the planar aromatic pyridine ring into a three-dimensional bicyclic architecture. This method not only achieves excellent yields, reported up to 80% in optimized conditions, but also demonstrates broad substrate tolerance, accommodating various electron-withdrawing and electron-donating groups on the aryl ring. Such versatility ensures that a wide library of analogs can be generated rapidly, accelerating the structure-activity relationship (SAR) studies essential for modern drug development.
Mechanistic Insights into Intramolecular Electrocyclization Dearomatization
The chemical elegance of this transformation lies in its mechanistic pathway, which bypasses the need for external oxidants or reductants by utilizing the internal energy of the conjugated system. The reaction initiates with the activation of the pyridine nitrogen, potentially facilitated by hydrogen bonding interactions with the hexafluoroisopropanol solvent, which acts as a strong hydrogen bond donor. This interaction increases the electrophilicity of the pyridine ring, making it susceptible to nucleophilic attack by the enol oxygen of the side chain. The subsequent electrocyclization step involves a concerted reorganization of pi-electrons, leading to the formation of the new carbon-nitrogen bond and the loss of aromaticity in the pyridine ring. This dearomatization is the driving force that locks the molecule into the rigid 3,4-dihydro-2H-quinolizin-2-one conformation. The specificity of this reaction is governed by the stereochemistry of the starting dienoate, where the (2Z,4E) configuration is critical for aligning the reactive orbitals correctly for ring closure. Understanding this mechanistic nuance is vital for process chemists aiming to replicate or optimize the synthesis for larger batches.
From an impurity control perspective, the mildness of the reaction conditions plays a pivotal role in ensuring product quality. Harsh acidic or basic conditions often lead to hydrolysis of the ester moiety or polymerization of the conjugated diene system, generating difficult-to-remove byproducts. By operating at room temperature in a non-nucleophilic fluorinated alcohol solvent, the process minimizes these side reactions, resulting in a cleaner crude reaction mixture. The solvent choice is particularly strategic; hexafluoroisopropanol not only stabilizes the transition state through specific solvation effects but also facilitates the proton transfer steps required for the tautomerization of the intermediate enol to the final ketone product. This intrinsic selectivity reduces the burden on downstream purification units, allowing for simpler chromatographic separation or even crystallization in some cases. For R&D directors, this implies a more predictable impurity profile, which simplifies the analytical validation required for regulatory filings and ensures a consistent supply of high-purity material for biological testing.
How to Synthesize 3,4-Dihydro-2H-quinolizin-2-one Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to solvent quality and reaction monitoring to ensure reproducibility. The protocol outlined in the patent emphasizes the use of dry reaction vessels and high-purity reagents to prevent premature hydrolysis of the sensitive dienoate starting material. The reaction progress is typically monitored using thin-layer chromatography (TLC), looking for the disappearance of the starting material spot and the emergence of a new product spot with distinct polarity. Once the reaction reaches completion, usually after a prolonged stirring period of several days to ensure full conversion, the workup involves a straightforward removal of the volatile solvent under reduced pressure. The resulting residue is then subjected to column chromatography, utilizing gradient elution with petroleum ether and ethyl acetate to isolate the pure orange solid product. Detailed standardized synthesis steps see the guide below.
- Dissolve the 2-pyridine acetate derivative substrate (II) in hexafluoroisopropanol solvent to achieve a molar concentration of approximately 0.1 mol/L.
- Stir the reaction mixture at room temperature for approximately 7 days to allow the intramolecular electrocyclization dearomatization to proceed to completion.
- Remove the solvent under reduced pressure and purify the resulting residue via column chromatography using petroleum ether and ethyl acetate mixtures.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthetic route offers tangible benefits that extend beyond the laboratory bench, directly impacting the bottom line and operational resilience. The elimination of expensive transition metal catalysts represents a significant cost avoidance strategy, as these metals often constitute a major portion of the raw material bill for complex heterocycle synthesis. Furthermore, the absence of metals removes the need for specialized scavenging resins and extensive testing for residual metals, streamlining the quality control workflow and reducing the overall cycle time from synthesis to release. The use of room temperature conditions also translates to lower energy consumption, as there is no requirement for heating mantles, oil baths, or cryogenic chillers, contributing to a greener and more cost-effective manufacturing process. These factors collectively enhance the economic feasibility of producing these intermediates at a commercial scale, making them accessible for broader application in drug discovery pipelines.
- Cost Reduction in Manufacturing: The process achieves substantial cost savings by utilizing a catalyst-free system or inexpensive organic acids, thereby eliminating the procurement costs associated with precious metal catalysts like palladium or rhodium. Additionally, the simplified workup procedure reduces the consumption of silica gel and solvents during purification, further lowering the variable costs per kilogram of product. The high atom economy of the intramolecular cyclization ensures that most of the starting material mass is incorporated into the final product, minimizing waste disposal fees. By avoiding complex protection and deprotection steps often required in alternative routes, the overall number of unit operations is reduced, leading to lower labor and overhead costs. This efficient use of resources allows for a more competitive pricing structure for the final API intermediate, providing a strategic advantage in price-sensitive markets.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, specifically the 2-pyridine acetate derivatives, are structurally simple and can be sourced from multiple global suppliers, mitigating the risk of single-source dependency. The robustness of the reaction conditions means that the process is less sensitive to minor fluctuations in temperature or reagent quality, ensuring consistent batch-to-batch performance even when scaling up. This reliability is crucial for maintaining continuous production schedules and meeting tight delivery deadlines for pharmaceutical clients. Moreover, the stability of the intermediates and the final product under ambient conditions simplifies logistics and storage requirements, reducing the need for specialized cold chain transportation. This logistical flexibility enhances the overall agility of the supply chain, allowing for quicker response times to fluctuating market demands.
- Scalability and Environmental Compliance: Scaling this reaction from gram to kilogram quantities is straightforward due to the lack of exothermic hazards associated with strong oxidants or reactive metals. The use of hexafluoroisopropanol, while a specialized solvent, allows for efficient recovery and recycling through distillation, aligning with green chemistry principles and reducing the environmental footprint of the manufacturing process. The mild reaction conditions also minimize the generation of hazardous byproducts, simplifying wastewater treatment and ensuring compliance with increasingly stringent environmental regulations. The ability to run the reaction in standard glass-lined or stainless steel reactors without the need for exotic materials of construction further facilitates rapid technology transfer from R&D to production. This scalability ensures that the supply of these critical intermediates can grow in tandem with the clinical development of the drug candidates that utilize them.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of these quinolizinone derivatives, based on the detailed data provided in the patent documentation. Understanding these aspects helps stakeholders make informed decisions about integrating this technology into their development pipelines. The answers reflect the specific advantages of the disclosed method over prior art, focusing on yield, purity, and operational simplicity. Clients are encouraged to review these points when evaluating the feasibility of this route for their specific project needs.
Q: What makes the synthesis of 3,4-dihydro-2H-quinolizin-2-one compounds challenging?
A: Traditional methods for constructing nitrogen-containing heterocycles often struggle with regioselectivity and harsh reaction conditions. Specifically, forming the 3,4-dihydro-2H-quinolizin-2-one skeleton has historically been difficult due to the stability of the aromatic pyridine ring, requiring complex multi-step sequences or expensive catalysts to achieve dearomatization.
Q: Does this novel method require expensive transition metal catalysts?
A: No, one of the significant advantages of this patented process is that it can proceed efficiently without any catalyst. While acids like p-toluenesulfonic acid can be used, the reaction achieves optimal yields (up to 80%) in hexafluoroisopropanol without added catalysts, significantly reducing raw material costs and metal contamination risks.
Q: Is this synthesis route suitable for large-scale manufacturing?
A: Yes, the process operates at room temperature and utilizes standard organic solvents, eliminating the need for cryogenic cooling or high-pressure equipment. This mild operational profile, combined with simple workup procedures involving solvent removal and column chromatography, makes it highly amenable to commercial scale-up for API intermediate production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4-Dihydro-2H-quinolizin-2-one Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this novel synthetic methodology in accelerating the discovery of new therapeutic agents. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can seamlessly transition from early-stage research to full-scale manufacturing. Our facilities are equipped with state-of-the-art rigorous QC labs capable of handling complex heterocyclic structures, guaranteeing that every batch meets stringent purity specifications required by global regulatory bodies. We understand that time-to-market is critical, and our team is prepared to optimize this room-temperature cyclization process to maximize throughput while maintaining the highest quality standards. By leveraging our expertise in process chemistry and supply chain management, we can help you secure a stable and cost-effective supply of these valuable intermediates.
We invite you to engage with our technical procurement team to discuss how this technology can be tailored to your specific drug discovery needs. Whether you require a Customized Cost-Saving Analysis for your current supply chain or need specific COA data to validate the quality of our materials, we are ready to provide comprehensive support. Our commitment to transparency and technical excellence ensures that you receive not just a chemical product, but a strategic partnership that drives your project forward. Contact us today to request route feasibility assessments and discover how we can support your journey from molecule to medicine with reliability and precision.