Advanced Two-Step Synthesis of 5-Fluorocytosine for Scalable API Manufacturing
The pharmaceutical industry constantly seeks robust synthetic routes for critical antiviral and antitumor intermediates, and the methodology disclosed in patent CN110615767B represents a significant leap forward in the production of 5-fluorocytosine. This specific patent details a streamlined, two-step synthesis that fundamentally alters the risk profile and cost structure associated with manufacturing this vital pyrimidine base. By utilizing N4-acyl protected cytosine as the starting material and employing organic carboxylic acids as solvents, the process successfully circumvents the severe equipment corrosion and safety hazards traditionally linked to liquid hydrogen fluoride protocols. The resulting 5-fluorocytosine achieves exceptional purity levels, consistently demonstrating HPLC purity greater than 99.9 percent in experimental trials, which is a critical specification for downstream API synthesis. For global supply chain leaders, this innovation offers a pathway to more reliable sourcing of high-quality nucleoside intermediates while mitigating the environmental and safety liabilities of legacy fluorination chemistries.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 5-fluorocytosine has been plagued by significant technical and safety hurdles that complicate large-scale manufacturing and inflate production costs. Conventional literature often describes routes utilizing liquid hydrogen fluoride as both a solvent and fluorinating agent, a practice that necessitates specialized, corrosion-resistant reactor vessels and imposes rigorous safety protocols due to the extreme toxicity and reactivity of HF. Alternative methods reported by researchers such as Morris J. Robins involve the use of expensive and environmentally harmful fluorine reagents like trifluorooxyborone at cryogenic temperatures of minus 78 degrees Celsius, which drastically increases energy consumption and operational complexity. Furthermore, routes starting from 2,5-difluoro-4-chloropyrimidine face supply chain bottlenecks because the starting material is difficult to prepare and lacks broad market availability. These legacy processes often suffer from impurity profiles that include hydroxyl-substituted by-products, requiring extensive and yield-reducing purification steps to meet pharmaceutical grade standards.
The Novel Approach
In stark contrast to these hazardous and costly legacy methods, the novel approach detailed in the patent introduces a protective group strategy that enables direct fluorination under remarkably mild and safe conditions. By employing N4-acyl cytosine derivatives, such as N4-acetylcytosine or N4-benzoylcytosine, the synthesis effectively masks the amino group, preventing unwanted side reactions during the fluorination step. The reaction proceeds smoothly in common organic carboxylic acids like glacial acetic acid or formic acid at moderate temperatures ranging from 40 to 50 degrees Celsius, eliminating the need for cryogenic cooling or aggressive HF handling. This strategic modification not only simplifies the operational workflow but also inherently suppresses the formation of 5-fluorouracil impurities, leading to a much cleaner reaction profile. The subsequent removal of the protecting group is achieved through a straightforward ammonolysis reaction in an alcohol solution, yielding the final product with high efficiency and minimal waste generation.
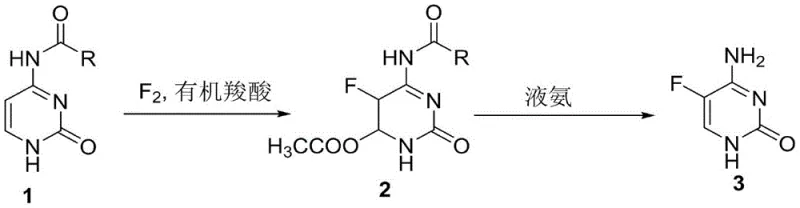
Mechanistic Insights into Electrophilic Fluorination with N-Protection
The core chemical innovation of this process lies in the electrophilic fluorination of the pyrimidine ring, facilitated by the electronic modulation provided by the N4-acyl protecting group. In the first step, fluorine gas, typically diluted with nitrogen for safety, acts as the electrophile attacking the electron-rich C5 position of the cytosine ring. The presence of the acyl group on the N4 nitrogen atom reduces the nucleophilicity of the exocyclic amine, thereby directing the fluorination selectively to the carbon ring rather than causing N-fluorination or oxidative degradation. This selectivity is paramount for maintaining the structural integrity of the pyrimidine base and ensuring that the fluorine atom is installed exclusively at the 5-position. The use of organic carboxylic acids as solvents further stabilizes the reaction intermediates and provides a proton source that aids in the rearomatization of the ring following the addition of fluorine, driving the reaction to completion without the need for harsh Lewis acids.
Furthermore, the mechanistic advantage extends to the impurity control profile, which is a primary concern for R&D directors overseeing API intermediate quality. In unprotected cytosine fluorination, the acidic environment often promotes the hydrolysis of the C4-amino group to a hydroxyl group, resulting in the formation of 5-fluorouracil, a structurally similar but pharmacologically distinct impurity that is difficult to separate. The N4-acyl protection renders the C4 position resistant to hydrolysis under the reaction conditions, effectively locking the amino functionality in place until the dedicated deprotection step. During the second stage, treatment with liquid ammonia in methanol or ethanol facilitates a nucleophilic attack on the carbonyl of the acyl group, cleaving the amide bond and regenerating the free amine. This two-stage separation of fluorination and deprotection ensures that the final 5-fluorocytosine is obtained with a purity profile that exceeds 99.9 percent, significantly reducing the burden on downstream purification processes.
How to Synthesize 5-Fluorocytosine Efficiently
The synthesis protocol outlined in the patent provides a clear and reproducible pathway for producing high-purity 5-fluorocytosine suitable for pharmaceutical applications. The process begins with the dissolution of the N4-acyl protected cytosine starting material in a selected organic carboxylic acid solvent, followed by the controlled introduction of fluorine gas at elevated temperatures. After the fluorination is complete, the solvent is recovered, and the intermediate is precipitated using methanol, allowing for easy isolation via filtration. The isolated intermediate is then subjected to ammonolysis in an alcohol-ammonia mixture to remove the protecting group, followed by aqueous refinement to yield the final crystalline product. For detailed operational parameters and specific stoichiometric ratios, please refer to the standardized synthesis guide below.
- React N4-acyl cytosine with fluorine gas in an organic carboxylic acid solvent at 40-50°C to form the fluorinated intermediate.
- Treat the intermediate with a liquid ammonia/alcohol solution at 40-50°C to remove the acyl protecting group.
- Refine the crude product using water crystallization to achieve high-purity 5-fluorocytosine (>99.9%).
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this patented methodology offers substantial strategic advantages by decoupling production from hazardous and scarce reagents. The elimination of liquid hydrogen fluoride and exotic fluorinating agents like trifluorooxyborone removes significant cost drivers associated with specialized storage, handling, and disposal of dangerous chemicals. By shifting to common solvents like glacial acetic acid and formic acid, the process leverages widely available commodity chemicals, ensuring stable pricing and consistent supply continuity even during market fluctuations. This transition also drastically simplifies the equipment requirements, allowing manufacturers to utilize standard glass-lined or stainless steel reactors instead of expensive Hastelloy or Monel vessels required for HF processing, leading to significant capital expenditure savings.
- Cost Reduction in Manufacturing: The economic benefits of this route are driven primarily by the substitution of expensive and hazardous reagents with cost-effective alternatives. By avoiding the use of continuous flow equipment and cryogenic systems, the process reduces both energy consumption and equipment depreciation costs. The high selectivity of the reaction minimizes the loss of valuable starting materials to by-products, thereby improving the overall mass balance and reducing the cost per kilogram of the active intermediate. Additionally, the simplified workup procedure, which involves straightforward precipitation and filtration, lowers labor and utility costs associated with complex chromatographic separations.
- Enhanced Supply Chain Reliability: The reliance on N4-acyl cytosine derivatives and standard organic acids creates a more resilient supply chain less susceptible to disruptions. Unlike routes dependent on hard-to-source precursors like 2,5-difluoro-4-chloropyrimidine, the raw materials for this process are commercially abundant and produced by multiple global suppliers. This diversity in sourcing options mitigates the risk of single-supplier dependency and ensures that production schedules can be maintained without interruption. The robustness of the chemistry also means that batch-to-batch variability is minimized, providing downstream API manufacturers with a consistent and reliable feedstock for their own synthesis campaigns.
- Scalability and Environmental Compliance: The mild reaction conditions and absence of highly toxic reagents make this process inherently scalable and environmentally compliant. Operating at temperatures between 40 and 50 degrees Celsius reduces the thermal load on the facility and eliminates the need for energy-intensive cooling systems. The waste stream is significantly less hazardous compared to HF-based processes, simplifying effluent treatment and reducing the environmental footprint of the manufacturing site. This alignment with green chemistry principles not only facilitates regulatory approval but also enhances the corporate sustainability profile of the manufacturing partner, a key consideration for modern pharmaceutical supply chains.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These answers are derived directly from the experimental data and technical specifications provided in the patent documentation to ensure accuracy and relevance for potential partners. Understanding these details is crucial for evaluating the feasibility of integrating this technology into existing production lines or new facility designs.
Q: Why is N4-acyl protection critical in this synthesis route?
A: The N4-acyl protection prevents the formation of hydroxyl by-products, specifically 5-fluorouracil, which typically occurs when unprotected cytosine is subjected to acidic fluorination conditions. This ensures the final API intermediate maintains exceptional purity levels exceeding 99.9%.
Q: What are the safety advantages over traditional liquid HF methods?
A: Traditional methods often utilize liquid hydrogen fluoride, which causes severe equipment corrosion and poses significant handling hazards. This novel process replaces hazardous HF with common organic carboxylic acids like glacial acetic acid, drastically improving operational safety and reducing maintenance costs.
Q: Can this process be scaled for industrial production?
A: Yes, the process utilizes readily available raw materials and mild reaction conditions (40-50°C), making it highly suitable for large-scale commercial manufacturing without the need for specialized continuous flow equipment or cryogenic temperatures.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Fluorocytosine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-purity intermediates in the development of life-saving antiviral and antitumor medications. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial reality is seamless and efficient. We are committed to maintaining stringent purity specifications and operating rigorous QC labs to guarantee that every batch of 5-fluorocytosine meets the exacting standards required by global regulatory bodies. Our infrastructure is designed to handle complex fluorination chemistries safely, leveraging the latest advancements in process safety and environmental protection.
We invite pharmaceutical partners to collaborate with us to leverage this advanced synthesis technology for their supply chains. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to reach out today to obtain specific COA data and comprehensive route feasibility assessments that demonstrate how our optimized manufacturing processes can enhance your product competitiveness and supply security.