Advanced Two-Step Synthesis of 5-Fluorocytosine for Commercial API Production
Introduction to Next-Generation 5-Fluorocytosine Manufacturing
The pharmaceutical industry continuously seeks robust, safe, and cost-effective pathways for synthesizing critical antiviral and antitumor intermediates. A pivotal advancement in this domain is detailed in patent CN110615767B, which discloses a novel method for synthesizing 5-fluorocytosine, a key precursor for medications such as capecitabine and lamivudine. This technology represents a significant departure from hazardous legacy processes, offering a streamlined two-step reaction sequence that prioritizes operator safety and environmental compliance without compromising on yield or quality. By leveraging an N4-acyl protection strategy, the process effectively mitigates the severe equipment corrosion and toxicity issues associated with traditional hydrogen fluoride routes.
For R&D directors and procurement specialists, the implications of this patent are profound. It offers a viable alternative to expensive fluorinating reagents and complex continuous flow setups, utilizing instead common organic carboxylic acids and direct fluorine gas. The result is a high-purity product, consistently achieving over 99.9% purity by HPLC, which drastically reduces downstream purification costs. As a reliable pharmaceutical intermediate supplier, understanding these mechanistic advantages is crucial for securing a stable supply chain for next-generation nucleoside analogues.
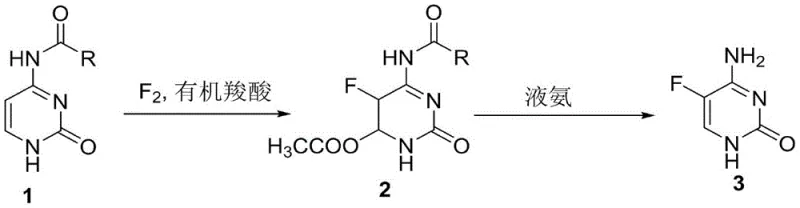
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of 5-fluorocytosine has been plagued by significant safety hazards and equipment corrosion issues, primarily due to the reliance on anhydrous hydrogen fluoride (HF) as both a solvent and a fluorinating agent. Literature reports, such as those by Tako Takahara, describe processes where cytosine is directly fluorinated in liquid HF, necessitating specialized corrosion-resistant reactors and posing extreme risks to personnel. Furthermore, alternative routes employing reagents like trifluorooxyborone or trichlorofluoromethane operate at cryogenic temperatures (-78°C), leading to prohibitive energy costs and the use of environmentally damaging ozone-depleting substances. Other methods involving 2,5-difluoro-4-chloropyrimidine suffer from the scarcity and high cost of starting materials, making them unsuitable for large-scale commercial adoption.
The Novel Approach
In stark contrast, the methodology disclosed in CN110615767B introduces a strategic N4-acyl protection group that fundamentally alters the reactivity of the cytosine ring, allowing for direct electrophilic fluorination under mild conditions. By using N4-acyl cytosine as the starting material and organic carboxylic acids (such as glacial acetic acid or formic acid) as solvents, the reaction proceeds efficiently at moderate temperatures between 40°C and 50°C. This approach not only eliminates the need for hazardous liquid HF but also simplifies the workup procedure, as the intermediate can be easily isolated by methanol precipitation. The subsequent deprotection step using ammonia in alcohol restores the aromatic system and removes the acyl group in a single operation, delivering the final API intermediate with exceptional purity.
Mechanistic Insights into N4-Acyl Protected Electrophilic Fluorination
The core innovation of this synthesis lies in the electronic modulation of the pyrimidine ring via N4-acylation. In unprotected cytosine, the amino group is highly nucleophilic and susceptible to side reactions or oxidation under harsh fluorination conditions. By converting the amino group into an amide (N4-acyl), the electron density is carefully tuned to favor electrophilic attack by fluorine gas specifically at the C5 position. The reaction mechanism involves the addition of fluorine across the C5-C6 double bond, facilitated by the organic acid solvent which likely stabilizes the transition state. This results in the formation of a stable intermediate, typically a 5-fluoro-6-acetoxy derivative, rather than immediate aromatization, which prevents over-fluorination or ring degradation.
Furthermore, this protection strategy plays a critical role in impurity control, specifically in suppressing the formation of 5-fluorouracil, a common and difficult-to-remove by-product in traditional acidic fluorination routes. In the absence of the N4-protecting group, acidic conditions can lead to the hydrolysis of the C4-amino group to a hydroxyl group. However, the acyl group shields the C4 position during the fluorination step. The final step, treatment with liquid ammonia in alcohol, serves a dual purpose: it acts as a base to eliminate the C6-substituent (e.g., acetoxy group) to restore aromaticity, and as a nucleophile to cleave the N4-amide bond, regenerating the free amino group. This tandem deprotection-aromatization ensures that the final product is chemically pure 5-fluorocytosine with minimal structural analogues.
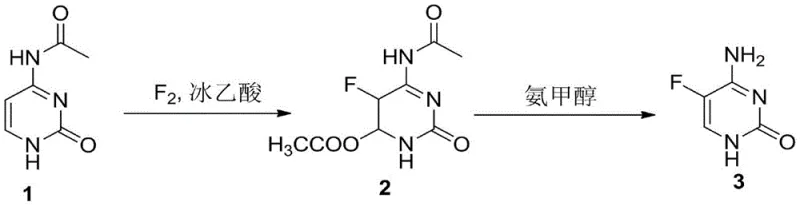
How to Synthesize 5-Fluorocytosine Efficiently
The practical implementation of this synthesis route is designed for ease of operation in standard chemical manufacturing facilities. The process begins with the suspension of N4-acyl cytosine in a selected organic carboxylic acid, followed by the controlled introduction of fluorine gas (often diluted with nitrogen for safety) at 40-50°C. Upon completion, the solvent is recovered, and the intermediate is precipitated using methanol. The second stage involves heating this intermediate in an ammonia-alcohol solution to effect the final transformation. For a comprehensive breakdown of the specific molar ratios, solvent choices, and isolation techniques validated in the patent examples, please refer to the standardized synthesis guide below.
- React N4-acyl cytosine with fluorine gas in an organic carboxylic acid solvent at 40-50°C to form the fluorinated intermediate.
- Treat the intermediate with a liquid ammonia/alcohol solution at 40-50°C to remove the protecting group and restore aromaticity.
- Purify the crude product via water refinement to achieve pharmaceutical grade purity exceeding 99.9%.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented process addresses several critical pain points in the supply chain of nucleoside intermediates. By shifting away from specialized continuous flow equipment and exotic fluorinating agents, manufacturers can utilize existing batch reactor infrastructure, significantly lowering capital expenditure barriers. The use of commodity chemicals like glacial acetic acid and fluorine gas ensures that raw material costs remain stable and predictable, insulating the supply chain from the volatility associated with niche reagents. Moreover, the high purity of the crude product reduces the burden on quality control laboratories and minimizes waste generation during recrystallization, contributing to a leaner and more sustainable manufacturing model.
- Cost Reduction in Manufacturing: The elimination of expensive fluorinating reagents such as trifluorooxyborone and the avoidance of cryogenic cooling systems leads to substantial operational cost savings. Additionally, the ability to recover and reuse organic acid solvents like acetic acid further enhances the economic efficiency of the process. By removing the need for costly heavy metal catalysts or complex purification columns often required to remove metal residues, the overall cost of goods sold (COGS) is significantly optimized, allowing for more competitive pricing in the global API market.
- Enhanced Supply Chain Reliability: The reliance on readily available starting materials, such as N4-acetylcytosine and common organic acids, ensures a robust supply chain that is less susceptible to disruptions. Unlike routes dependent on 2,5-difluoro-4-chloropyrimidine, which may have limited suppliers, the precursors for this method are produced by multiple chemical vendors globally. This diversification of the raw material base guarantees consistent production schedules and shorter lead times for high-purity pharmaceutical intermediates, enabling partners to meet tight project deadlines with confidence.
- Scalability and Environmental Compliance: The process operates at mild temperatures and atmospheric pressure, making it inherently safer and easier to scale from pilot plant to commercial production volumes. The absence of liquid hydrogen fluoride eliminates the need for specialized HF-resistant alloys and complex waste neutralization systems, thereby reducing the environmental footprint of the facility. This alignment with green chemistry principles not only simplifies regulatory compliance but also enhances the corporate sustainability profile of the manufacturing partner, a key factor for modern pharmaceutical procurement strategies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing a clear picture of the process capabilities and limitations for potential adopters.
Q: How does this method improve safety compared to traditional HF processes?
A: This method eliminates the need for anhydrous hydrogen fluoride (HF), a highly corrosive and toxic reagent. By using organic carboxylic acids like glacial acetic acid as solvents and fluorine gas directly, it significantly reduces equipment corrosion risks and operational hazards.
Q: What purity levels can be achieved with this synthesis route?
A: The patented process consistently yields 5-fluorocytosine with HPLC purity greater than 99.9%. The N4-acyl protection strategy effectively prevents the formation of hydroxyl by-products like 5-fluorouracil, ensuring a clean impurity profile suitable for API manufacturing.
Q: Is this process scalable for industrial production?
A: Yes, the process is designed for scalability. It utilizes common, inexpensive reagents and operates at mild temperatures (40-50°C). Unlike continuous flow methods requiring specialized equipment, this batch process can be easily adapted for large-scale commercial production ranging from hundreds of kilograms to multi-ton capacities.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Fluorocytosine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic methodologies to ensure the consistent supply of high-quality API intermediates. Our technical team has extensively analyzed the N4-acyl protection route described in CN110615767B and possesses the expertise to implement this chemistry effectively. We boast extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive material that meets stringent purity specifications. Our rigorous QC labs are equipped to verify the absence of critical impurities like 5-fluorouracil, guaranteeing that every batch of 5-fluorocytosine delivered is ready for downstream pharmaceutical synthesis.
We invite procurement managers and R&D leaders to collaborate with us to leverage this cost-effective and safe manufacturing route. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, ensuring that your supply chain for antiviral and antitumor intermediates is built on a foundation of technical excellence and commercial reliability.