Scalable Iron-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Advanced Drug Discovery
Scalable Iron-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Advanced Drug Discovery
The landscape of modern pharmaceutical research is increasingly dominated by the demand for complex heterocyclic scaffolds that offer superior metabolic stability and binding affinity. Among these, quinazolinone derivatives stand out as privileged structures found in numerous bioactive natural products and clinical drugs, exhibiting potent anti-cancer, anticonvulsant, and anti-inflammatory properties. A pivotal advancement in this domain is disclosed in patent CN111675662B, which details a robust and efficient preparation method for 2-trifluoromethyl substituted quinazolinone compounds. This technology addresses the critical need for introducing fluorine atoms into heterocyclic systems, a modification known to drastically improve the electronegativity, lipophilicity, and overall bioavailability of drug candidates. By leveraging a novel iron-catalyzed cyclization strategy, this method provides a streamlined pathway to access these valuable molecular architectures, overcoming many of the limitations associated with traditional synthetic routes.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinazolinones bearing trifluoromethyl functionalities has relied heavily on the cyclization of specific synthons such as anthranilamide, anthranilic acid, or isatoic anhydride with trifluoromethyl-containing reagents. While effective in certain contexts, these conventional methodologies are frequently plagued by severe operational constraints that hinder their utility in large-scale manufacturing. Common trifluoromethylating agents like trifluoroacetic anhydride or ethyl trifluoroacetate often necessitate harsh reaction conditions, including extreme temperatures or the use of strong, hazardous bases. Furthermore, these traditional routes frequently suffer from narrow substrate scope, meaning they fail to accommodate diverse functional groups without significant yield penalties. The reliance on expensive starting materials and the generation of difficult-to-remove byproducts further exacerbate the cost and complexity, making these methods less attractive for the cost-sensitive production of pharmaceutical intermediates.
The Novel Approach
In stark contrast to the cumbersome legacy techniques, the innovative protocol outlined in the patent utilizes readily available isatin derivatives and trifluoroethylimidoyl chloride as the primary building blocks. This strategic shift in retrosynthetic design allows for a direct and atom-economical construction of the quinazolinone core. The reaction is driven by a cheap and abundant iron catalyst, specifically ferric chloride, which operates effectively under relatively mild thermal conditions. This new approach not only simplifies the operational workflow by eliminating the need for exotic reagents but also demonstrates exceptional functional group tolerance. The ability to synthesize a wide array of substituted quinazolinones with high efficiency and yield represents a significant leap forward, enabling medicinal chemists to rapidly explore structure-activity relationships (SAR) without being bottlenecked by synthetic complexity.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The core of this technological breakthrough lies in the unique mechanistic pathway facilitated by the ferric chloride catalyst. The reaction initiates with the activation of the substrates, where the Lewis acidic nature of the iron species likely coordinates with the carbonyl oxygen of the isatin, increasing its electrophilicity. Simultaneously, the presence of sodium hydride promotes the formation of key carbon-nitrogen bonds between the trifluoroethylimidoyl chloride and the isatin nitrogen. This initial condensation leads to the formation of a transient trifluoroacetamidine intermediate. Subsequently, the iron catalyst plays a crucial role in mediating a decarbonylation event, followed by an intramolecular cyclization that closes the six-membered ring to form the stable quinazolinone scaffold. This cascade transformation is highly efficient, converting simple precursors into complex heterocycles in a single pot.
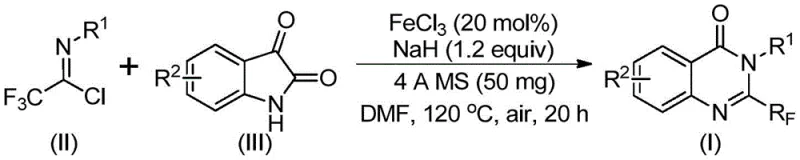
Beyond the primary transformation, the reaction conditions are meticulously optimized to ensure high purity and minimize the formation of side products. The use of 4A molecular sieves serves a dual purpose: acting as a desiccant to remove trace moisture that could deactivate the catalyst or hydrolyze sensitive intermediates, and potentially stabilizing the transition states. The tolerance of the system towards various substituents on both the aryl ring of the imidoyl chloride and the benzene ring of the isatin is remarkable. Whether electron-donating groups like methyl or methoxy, or electron-withdrawing groups like halogens and nitro groups are present, the catalytic cycle proceeds smoothly. This robustness suggests that the rate-determining steps are not significantly perturbed by electronic variations, providing a reliable platform for synthesizing diverse libraries of fluorinated heterocycles for drug discovery programs.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires adherence to specific procedural parameters to maximize yield and safety. The process begins with the precise weighing of ferric chloride and sodium hydride, which are added to a reaction vessel along with 4A molecular sieves. The choice of solvent is critical, with polar aprotic solvents like DMF proving most effective for dissolving the ionic intermediates and facilitating the reaction kinetics. The reaction profile involves a two-stage heating protocol, starting at a lower temperature to allow for initial mixing and intermediate formation, followed by heating to 120°C to drive the cyclization to completion. Detailed standardized operating procedures regarding stoichiometry, addition rates, and quenching protocols are essential for reproducibility.
- Mix ferric chloride, sodium hydride, 4A molecular sieves, trifluoroethylimidoyl chloride, and isatin derivative in an organic solvent like DMF.
- Stir the reaction mixture at 40°C for 8-10 hours, then heat to 120°C and continue reacting for 18-20 hours under air.
- Filter the mixture, mix with silica gel, and purify via column chromatography to isolate the final 2-trifluoromethyl substituted quinazolinone compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this iron-catalyzed methodology presents a compelling value proposition centered on cost optimization and supply security. The shift away from precious metal catalysts to earth-abundant iron salts fundamentally alters the cost structure of the synthesis. Iron catalysts are orders of magnitude cheaper than palladium or rhodium alternatives, and they do not carry the same geopolitical supply risks or price volatility. Moreover, the starting materials—isatin derivatives and aromatic amines used to make the imidoyl chlorides—are commodity chemicals produced on a massive global scale. This ensures a stable and continuous supply chain, reducing the risk of production delays caused by raw material shortages.
- Cost Reduction in Manufacturing: The economic benefits of this process are multifaceted, extending beyond just the price of the catalyst. By utilizing a one-pot synthesis that combines bond formation and cyclization, the number of unit operations is significantly reduced. Fewer isolation and purification steps translate directly into lower labor costs, reduced solvent consumption, and decreased waste disposal fees. The elimination of expensive ligands often required for transition metal catalysis further drives down the bill of materials. Additionally, the high conversion rates observed in this method mean that less raw material is wasted, maximizing the output per batch and improving the overall process mass intensity (PMI), a key metric for green and cost-effective manufacturing.
- Enhanced Supply Chain Reliability: Reliability in the supply of critical intermediates is paramount for maintaining uninterrupted drug production schedules. This synthesis route relies on chemically robust reagents that have long shelf lives and are widely available from multiple global suppliers. Unlike sensitive organometallic reagents that require specialized storage and handling, the reagents in this protocol are stable under standard conditions. This stability simplifies logistics and warehousing, allowing for larger inventory buffers without the risk of degradation. Consequently, manufacturers can respond more agilely to fluctuations in demand, ensuring that downstream API production is never compromised by upstream intermediate shortages.
- Scalability and Environmental Compliance: As regulatory pressures regarding environmental impact intensify, the sustainability of chemical processes becomes a key differentiator. This iron-catalyzed method aligns well with green chemistry principles by avoiding toxic heavy metals and minimizing waste generation. The simplicity of the workup, which typically involves filtration and standard chromatography, reduces the volume of hazardous waste streams. From a scalability perspective, the reaction conditions are mild enough to be safely managed in large reactors without requiring exotic high-pressure equipment. The demonstrated ability to scale from gram-level discovery to multi-kilogram production ensures that this technology can seamlessly support the entire lifecycle of a drug candidate, from early-stage clinical trials to commercial launch.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing a clear understanding of the method's capabilities and limitations for potential adopters.
Q: What are the primary advantages of using FeCl3 over precious metal catalysts for quinazolinone synthesis?
A: The use of ferric chloride (FeCl3) offers significant economic and operational advantages compared to traditional precious metal catalysts. Iron is an earth-abundant, non-toxic, and inexpensive metal, which drastically reduces the raw material costs associated with the catalytic system. Furthermore, iron catalysts often exhibit excellent functional group tolerance, allowing for the synthesis of complex heterocycles without the need for extensive protecting group strategies, thereby simplifying the overall synthetic route and improving process efficiency.
Q: How does the introduction of a trifluoromethyl group impact the biological activity of quinazolinone derivatives?
A: Introducing a trifluoromethyl group into the quinazolinone scaffold significantly enhances the physicochemical and pharmacological properties of the molecule. The high electronegativity and lipophilicity of the CF3 group improve metabolic stability and bioavailability, which are critical parameters for drug candidates. Additionally, the trifluoromethyl moiety can modulate the electronic distribution of the heterocyclic ring, potentially enhancing binding affinity to biological targets such as kinases or receptors involved in cancer and inflammation pathways.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the described method is highly amenable to scale-up due to its use of commercially available starting materials and robust reaction conditions. The protocol utilizes inexpensive reagents like isatin and avoids sensitive organometallic species that require strict inert atmospheres, as the reaction can proceed under air. The straightforward workup procedure involving filtration and standard column chromatography further supports its feasibility for manufacturing multi-kilogram quantities of high-purity intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in accelerating drug development timelines. Our team of expert chemists has extensively evaluated the FeCl3-catalyzed cyclization route and confirmed its potential for delivering high-purity 2-trifluoromethyl quinazolinones. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with consistency and precision. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch meets the exacting standards required for pharmaceutical applications.
We invite you to collaborate with us to leverage this advanced synthetic technology for your next project. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our optimized manufacturing processes can enhance your supply chain efficiency and reduce your overall cost of goods sold.