Scalable Synthesis of Afoxolaner Intermediates: Technical Breakthroughs for Commercial Manufacturing
The pharmaceutical and agrochemical industries continuously seek robust synthetic pathways that balance high purity with economic viability, particularly for complex veterinary actives like afoxolaner. Patent CN116143652A introduces a transformative preparation method for afoxolaner intermediates that addresses critical bottlenecks in the existing supply chain. This innovation focuses on optimizing the construction of the core naphthalene-amide scaffold, which is pivotal for the biological activity of the final isoxazoline compound. By re-engineering the amidation step and selecting widely available starting materials, the disclosed technology offers a compelling alternative to legacy processes that rely on costly, custom-made fragments. For R&D directors and procurement strategists, understanding this shift is essential for securing a stable supply of high-quality veterinary drug intermediates.
The structural integrity of afoxolaner relies heavily on the precise stereochemistry and purity of its precursors. The patent details a route that minimizes impurity generation during the early stages of synthesis, which is crucial for downstream processing. 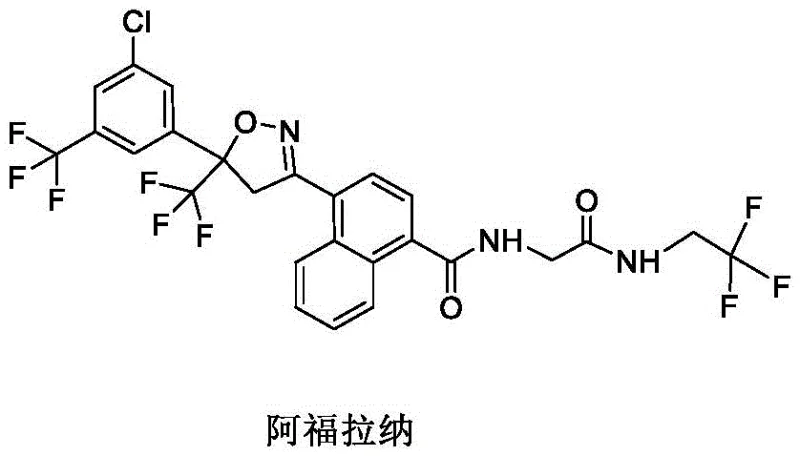
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methods, such as those disclosed in patent WO2009002809A2, have long plagued manufacturers with inefficiencies that drive up costs and extend lead times. The traditional route typically involves a lengthy sequence of reactions culminating in a difficult final butt-joint step that requires specialized insertion reactions in the presence of catalysts. A major drawback is the reliance on fragments that are not produced in large quantities industrially, necessitating custom synthesis or self-manufacturing of key building blocks like 2-amino-N-(2,2-trifluoroethyl)acetamide. This dependency creates significant supply chain vulnerabilities and inflates the cost of goods sold due to the low overall yield and complex purification requirements associated with these bespoke intermediates.
The Novel Approach
In stark contrast, the methodology outlined in CN116143652A streamlines the synthesis by utilizing cheap and easily accessible raw materials, fundamentally altering the cost structure of production. 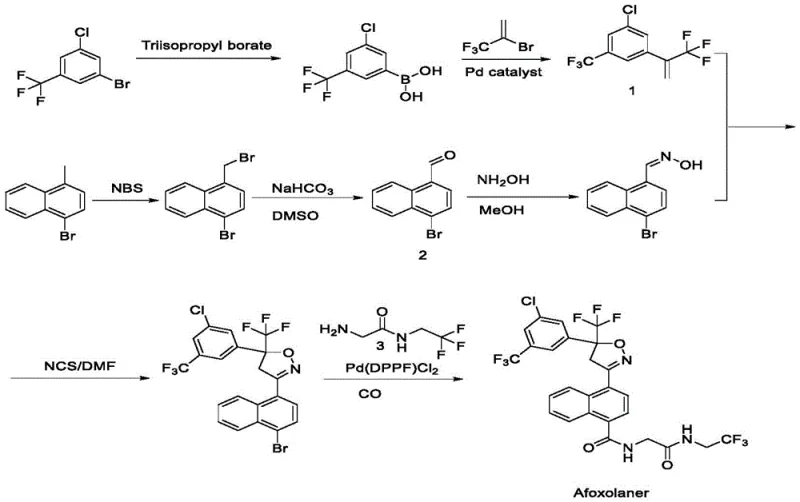
The new approach bypasses the need for expensive custom fragments by employing glycine ester derivatives and trifluoroethylamine hydrochloride, which are commodity chemicals with stable global supply chains. The reaction conditions are notably mild, operating safely within a temperature range of 0°C to 85°C, which reduces energy consumption and safety risks in the plant. Furthermore, the process is designed with industrial mass production in mind, demonstrating resilience against the 'amplification effect' where reaction efficiency typically drops when moving from lab bench to large reactors. This ensures consistent quality and yield regardless of batch size.
Mechanistic Insights into CDI-Mediated Amidation and Cyclization
The core of this technological advancement lies in the optimized amidation reaction between 5-acetyl-1-naphthoic acid and glycine ester derivatives. The mechanism utilizes N,N'-carbonyldiimidazole (CDI) as a condensing agent, activated specifically by the addition of methanesulfonic acid. This activation step is not merely catalytic but plays a stoichiometric role in ensuring the complete conversion of the carboxylic acid group. The molar ratio of methanesulfonic acid to the naphthoic acid derivative is carefully controlled at approximately 2:1, which protonates the intermediate and facilitates the nucleophilic attack by the amine. This precise control prevents the accumulation of unreacted starting materials, a common issue in large-scale amidations that can complicate downstream purification.
Following the formation of the amide intermediate, the synthesis proceeds through a condensation with trifluoroethylamine and subsequent reaction with a chloro-trifluoromethyl phenyl ketone to form the chalcone-like precursor. 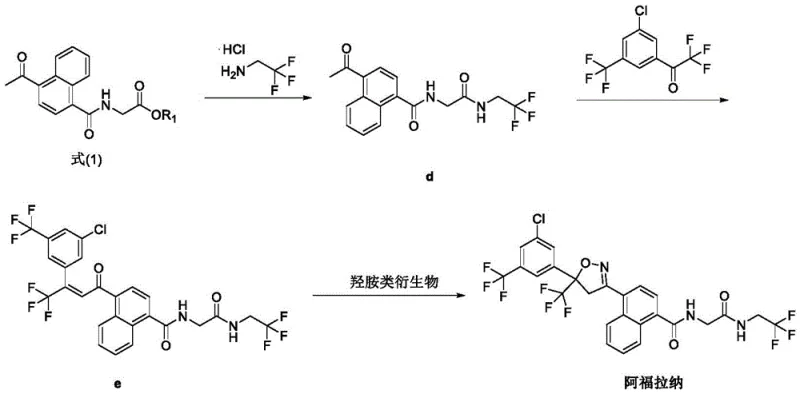
The final cyclization to form the isoxazoline ring is achieved using hydroxylamine derivatives under basic conditions. The choice of base, such as sodium hydroxide or DBU, and the solvent system, typically tetrahydrofuran or acetonitrile, is critical for controlling the regioselectivity of the ring closure. The patent highlights that maintaining the reaction temperature between 5°C and 60°C during this step minimizes side reactions and ensures the formation of the desired isomer with high chromatographic purity. This mechanistic precision is what allows the process to achieve yields significantly higher than prior art, directly translating to better resource utilization.
How to Synthesize Afoxolaner Intermediate Efficiently
The synthesis protocol described in the patent provides a clear roadmap for replicating these results in a GMP environment. The process begins with the activation of the naphthoic acid derivative followed by sequential coupling and cyclization steps. Each stage has been rigorously screened for solvent compatibility, temperature tolerance, and reagent equivalents to ensure robustness. For technical teams looking to implement this route, adherence to the specified molar ratios and temperature profiles is essential to replicate the high purity and yield reported in the examples. Detailed standardized synthesis steps are provided below to guide your process development team.
- React 5-acetyl-1-naphthoic acid with glycine ester derivatives using CDI and methanesulfonic acid to form the key amide intermediate.
- Condense the amide intermediate with trifluoroethylamine hydrochloride and subsequently with chloro-trifluoromethyl phenyl ketone.
- Perform final cyclization using hydroxylamine derivatives under basic conditions to close the isoxazoline ring.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the implications of this patented process extend far beyond simple chemistry; it represents a strategic opportunity to de-risk the supply of critical veterinary intermediates. By shifting away from custom-synthesized, high-cost fragments to commodity chemicals, the manufacturing cost profile is drastically improved. The elimination of expensive reagents like 2-amino-N-(2,2-trifluoroethyl)acetamide, which historically commanded exorbitant prices, removes a major cost driver from the bill of materials. This substitution allows for substantial cost savings that can be passed down the supply chain or reinvested into quality control measures.
- Cost Reduction in Manufacturing: The primary economic advantage stems from the replacement of proprietary, high-price starting materials with widely available glycine esters and trifluoroethylamine salts. This switch reduces the raw material cost to a fraction of the prior art, as the new reagents are produced at a global scale with competitive pricing. Additionally, the simplified purification process reduces solvent consumption and waste disposal costs, further enhancing the overall economic efficiency of the manufacturing campaign without compromising on the quality of the final active ingredient.
- Enhanced Supply Chain Reliability: Relying on commodity chemicals significantly mitigates the risk of supply disruptions. Unlike custom fragments that may have single-source suppliers or long lead times, glycine esters and common solvents like acetonitrile and dichloromethane are available from multiple vendors worldwide. This diversification ensures continuity of supply even during market fluctuations. Furthermore, the mild reaction conditions reduce the dependency on specialized equipment, allowing for more flexible production scheduling across different manufacturing sites.
- Scalability and Environmental Compliance: The process has been validated at the kilogram level, proving its suitability for commercial scale-up of complex veterinary drug intermediates. The use of methanesulfonic acid to overcome amplification effects ensures that yield and purity remain consistent as batch sizes increase to multi-ton scales. Moreover, the reduction in reaction steps and the use of less hazardous reagents contribute to a smaller environmental footprint, aligning with modern green chemistry principles and easing regulatory compliance burdens related to waste management and emissions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These answers are derived directly from the experimental data and beneficial effects reported in the patent documentation. They are intended to provide clarity on the feasibility and advantages of adopting this method for large-scale production.
Q: How does this new synthesis route improve upon the prior art WO2009002809A2?
A: The new route described in CN116143652A eliminates the need for expensive, custom-synthesized fragments like 2-amino-N-(2,2-trifluoroethyl)acetamide. By utilizing commercially available glycine esters and optimizing the amidation conditions with methanesulfonic acid, the process significantly reduces raw material costs and simplifies purification, overcoming the low yield and long route issues of the previous method.
Q: What specific technical challenges does the addition of methanesulfonic acid address?
A: The addition of methanesulfonic acid is critical for overcoming the 'amplification effect' observed during scale-up. In kilogram-level reactions, equipment changes often lead to incomplete raw material conversion. Methanesulfonic acid accelerates the reaction rate and ensures complete consumption of the starting naphthoic acid derivative, thereby maintaining high purity and yield even in large-scale industrial reactors.
Q: Is this synthesis method suitable for large-scale commercial production?
A: Yes, the patent explicitly validates the method at the kilogram level, demonstrating robustness against scale-up variables. The use of common solvents like acetonitrile and dichloromethane, along with mild reaction temperatures ranging from 0°C to 85°C, ensures that the process is safe, environmentally manageable, and readily adaptable to multi-ton annual production capacities.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Afoxolaner Intermediate Supplier
The technical potential of the route described in CN116143652A is immense, offering a pathway to high-purity intermediates that meet the stringent demands of the veterinary pharmaceutical market. NINGBO INNO PHARMCHEM stands ready to leverage this chemistry as part of our comprehensive CDMO services. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from lab to plant is seamless. Our rigorous QC labs and commitment to stringent purity specifications guarantee that every batch of afoxolaner intermediate we produce adheres to the highest international standards.
We invite you to discuss how this optimized synthesis can enhance your supply chain resilience and cost structure. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can support your long-term business goals.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →