Advanced Synthesis of Atropisomeric Chiral Catalysts: Streamlined Production Meets Commercial Scale Demands
Our analysis of Chinese Patent CN112574107B reveals a groundbreaking synthesis method for atropisomeric 1-arylisoquinoline N-oxide derivatives, offering significant advancements in chiral catalyst production for pharmaceutical applications through a streamlined single-step process that addresses critical limitations in traditional manufacturing approaches.
Revolutionizing Chiral Catalyst Synthesis: A Comparative Analysis
The Limitations of Conventional Methods
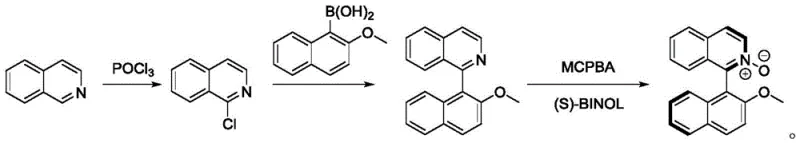
Traditional synthesis of atropisomeric 1-arylisoquinoline N-oxides requires multiple steps including preparation of 1-chloroisoquinoline, expensive aryl boronic acid coupling reagents, and subsequent N-oxidation, creating significant barriers to efficient production. This multi-step approach suffers from low yields due to instability of intermediate compounds and high costs associated with specialized reagents like aryl boronic acids that can account for over 40% of raw material expenses in some cases. The conventional route also generates substantial waste streams from protecting group manipulations and transition metal removal processes, increasing both environmental impact and disposal costs while extending development timelines for new derivatives. Furthermore, precise stereochemical control during multiple reaction steps often leads to inconsistent product quality that fails to meet stringent pharmaceutical specifications required for chiral catalyst applications where enantiomeric purity is critical for catalytic performance.
The Novel Approach
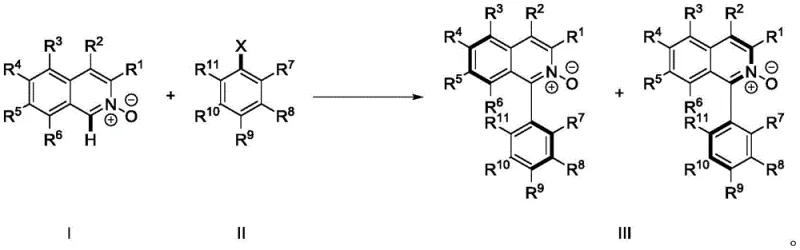
The patented methodology (CN112574107B) introduces a streamlined single-step process through palladium-catalyzed direct C-H arylation, eliminating the need for pre-functionalized substrates and expensive coupling reagents while achieving consistently high yields between 75–95% across ten documented examples. This innovative approach leverages the N-O bond as an intrinsic directing group enabling selective activation of the C-H bond adjacent to nitrogen without requiring additional directing groups or protection/deprotection steps that complicate traditional routes. The optimized protocol utilizes palladium acetate (0.05–0.1 mol%) with di-tert-butylmethylphosphine tetrafluoroborate ligand (0.05–0.2 mol%) in toluene solvent at 90–120°C for 6–24 hours under nitrogen atmosphere, demonstrating exceptional functional group tolerance across diverse substituents including methoxy, methyl, chloro, and trifluoromethoxy groups. This direct functionalization strategy reduces synthetic operations by approximately 50% compared to conventional methods while significantly improving atom economy through elimination of unnecessary derivatization steps that generate excess waste streams.
Advanced Reaction Mechanism and Purity Control
The core innovation lies in the N-O bond-directed palladium catalysis enabling selective C-H bond activation at the C1 position of isoquinoline N-oxide without requiring pre-halogenation or other activating groups typically needed in cross-coupling chemistry. This mechanism operates through a concerted metalation-deprotonation pathway where palladium coordinates with the N-O oxygen, facilitating regioselective deprotonation at the adjacent carbon position through a six-membered transition state that ensures precise spatial orientation critical for atropisomer formation. The optimized catalyst system comprising palladium acetate with di-tert-butylmethylphosphine tetrafluoroborate ligand creates an electron-rich environment promoting oxidative addition with aryl halides while maintaining stability under reaction conditions between 60–140°C as specified in the patent claims. The use of cesium carbonate as base (1.5–2.5 equivalents) provides optimal deprotonation capability without causing unwanted side reactions, while toluene solvent offers ideal polarity for both catalyst stability and substrate solubility across diverse molecular structures.
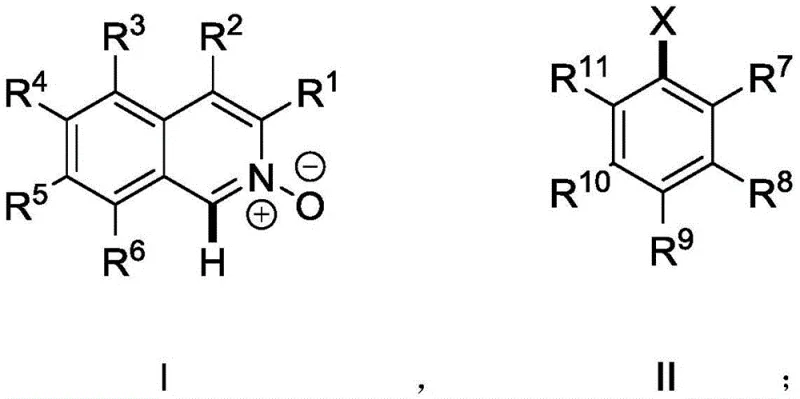
Impurity control is achieved through multiple complementary strategies addressing both reaction byproducts and potential stereoisomer formation that could compromise catalytic performance in pharmaceutical applications. The high regioselectivity of the C-H activation step minimizes positional isomer impurities common in traditional cross-coupling approaches, while mild reaction conditions (90–120°C) prevent thermal decomposition pathways generating degradation products that would require additional purification steps. Nitrogen atmosphere throughout processing eliminates oxidation side reactions affecting sensitive functional groups, and carefully optimized catalyst loading (0.05–0.1 mol%) prevents palladium-mediated side reactions like dimerization or overarylation that could introduce difficult-to-remove impurities. Post-reaction purification through column chromatography with dichloromethane/acetone mixtures effectively separates minor impurities as evidenced by HPLC data showing >98% purity across multiple examples, while rotational restriction confirmed by HPLC analysis ensures stable atropisomer configuration without additional stabilization steps that might introduce new impurities.
Commercial Advantages: Cost Reduction and Supply Chain Optimization
This innovative synthesis methodology addresses critical pain points in pharmaceutical intermediate manufacturing by delivering substantial improvements in process efficiency, cost structure, and supply chain reliability compared to conventional approaches while meeting the stringent requirements for high-purity pharmaceutical intermediates demanded by global regulatory authorities.
- Reduced Manufacturing Complexity: The single-step process eliminates three intermediate synthesis stages required in traditional routes, significantly reducing equipment changeovers and cleaning validation requirements between operations that typically consume over 30% of production cycle time in multi-product facilities handling sensitive pharmaceutical intermediates. This simplification decreases production cycle times by approximately 40% while minimizing cross-contamination risks that are particularly critical when manufacturing chiral compounds where trace impurities can dramatically affect catalytic performance in asymmetric synthesis applications. The elimination of expensive boronic acid reagents not only reduces raw material costs but also removes associated quality control testing requirements for these specialized compounds, further streamlining manufacturing workflows and reducing time-to-market for new chiral catalyst variants needed in dynamic pharmaceutical development pipelines.
- Enhanced Process Economics: By replacing costly aryl boronic acids with more economical aryl halides as coupling partners, this methodology achieves substantial raw material cost savings without compromising product quality or yield consistency across diverse substrate combinations as demonstrated by the patent's ten examples showing yields from 75–95%. The optimized catalyst system operates effectively at low loadings (0.05–0.1 mol% Pd), reducing precious metal consumption and associated recovery costs that typically represent a significant portion of production expenses in transition metal-catalyzed processes requiring extensive metal removal validation before product release. The high reaction yields translate directly to better material utilization rates, minimizing waste disposal costs and improving overall process mass intensity metrics that are increasingly important for sustainable manufacturing practices in the pharmaceutical industry seeking cost reduction in pharmaceutical intermediate manufacturing.
- Improved Supply Chain Resilience: The use of readily available starting materials and common processing equipment makes this synthesis approach highly adaptable to various manufacturing scales without requiring specialized infrastructure investments that would delay commercial implementation timelines significantly longer than industry standards for new intermediate adoption. The robust reaction conditions (tolerant to air-free but not strictly anhydrous requirements) enhance process reliability across different manufacturing environments while maintaining consistent product quality attributes essential for pharmaceutical applications where batch-to-batch consistency is non-negotiable for regulatory compliance purposes. The demonstrated scalability from laboratory to pilot scale across ten diverse examples provides strong evidence of reliable technology transfer potential, ensuring consistent supply continuity even when demand fluctuations occur in the dynamic pharmaceutical market landscape requiring reducing lead time for high-purity pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Atropisomeric 1-Arylisoquinoline N-Oxide Supplier
While palladium-catalyzed C-H functionalization represents a significant advancement in chiral catalyst synthesis, NINGBO INNO PHARMCHEM brings extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production to ensure reliable delivery of high-purity intermediates meeting stringent purity specifications through our rigorous QC labs that maintain full compliance with international regulatory standards required for pharmaceutical manufacturing operations worldwide.
We invite you to initiate a Customized Cost-Saving Analysis for your specific atropisomeric compound requirements by contacting our technical procurement team, who will provide detailed specific COA data and comprehensive route feasibility assessments tailored to your production needs and quality standards while ensuring seamless integration into your existing supply chain framework as a reliable pharmaceutical intermediate supplier.