Scalable Palladium-Catalyzed Synthesis of 3-Arylquinolin-2(1H)-one Derivatives for Pharmaceutical Applications
The pharmaceutical industry continuously seeks robust and efficient synthetic routes for heterocyclic scaffolds that serve as critical cores in drug discovery and development. A significant advancement in this domain is detailed in patent CN113045489B, which discloses a novel preparation method for 3-arylquinolin-2(1H)one derivatives. These quinolinone structures are privileged motifs found in numerous bioactive natural products and therapeutic agents, ranging from antibiotics and antiplatelet drugs to antitumor agents and receptor antagonists. The traditional reliance on multi-step syntheses or harsh reaction conditions has often hindered the rapid exploration of chemical space around this scaffold. However, the methodology described in this patent introduces a streamlined palladium-catalyzed aminocarbonylation strategy that fundamentally shifts the paradigm for accessing these valuable intermediates. By leveraging benzisoxazole as a unique dual-purpose reagent acting as both the nitrogen and formyl source, this innovation addresses key bottlenecks in synthetic efficiency and operational simplicity. As a reliable pharmaceutical intermediate supplier, understanding such technological leaps is crucial for maintaining a competitive edge in the global supply chain.
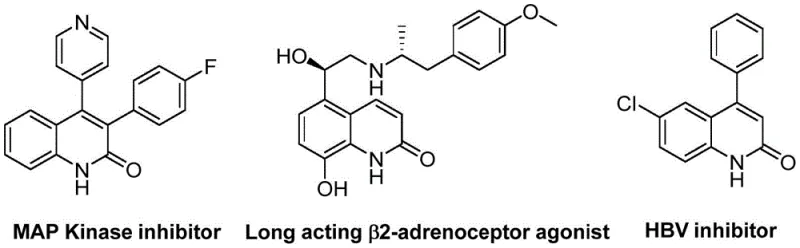
The significance of this technology extends beyond mere academic interest; it represents a tangible opportunity for cost reduction in API manufacturing. The ability to construct the quinolinone core in a single pot from readily available precursors like benzyl chlorides and benzisoxazoles reduces the cumulative waste and energy consumption associated with stepwise constructions. For R&D directors focused on purity and impurity profiles, the high selectivity of this catalytic system minimizes the formation of difficult-to-remove byproducts. Furthermore, the broad substrate scope demonstrated in the patent suggests that this method can be universally applied to generate diverse libraries of analogs, accelerating the lead optimization phase in drug discovery programs. This report delves into the mechanistic intricacies and commercial implications of adopting this state-of-the-art synthesis for commercial scale-up of complex pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinolin-2(1H)one derivatives has relied heavily on classical named reactions such as the Vilsmeier-Haack, Knorr, and Friedlander condensations. While these methods are well-established, they frequently suffer from significant drawbacks that impact their viability for modern, green chemistry-focused manufacturing. For instance, the Vilsmeier-Haack reaction often requires the use of toxic phosphoryl chloride and generates substantial amounts of acidic waste, posing environmental and safety challenges that increase disposal costs. Similarly, the Knorr and Friedlander reactions typically demand high temperatures and strong acidic or basic conditions, which can be incompatible with sensitive functional groups present on advanced intermediates. This lack of chemoselectivity often necessitates additional protection and deprotection steps, drastically increasing the overall step count and reducing the final overall yield. Moreover, transition metal-catalyzed improvements to these classical methods have sometimes required expensive ligands or hazardous carbon monoxide gas sources, creating barriers to safe implementation in standard pilot plants. These limitations collectively result in longer lead times for high-purity pharmaceutical intermediates and inflated production costs that erode profit margins.
The Novel Approach
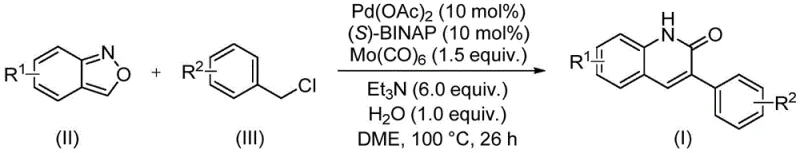
In stark contrast, the novel approach outlined in the patent data utilizes a sophisticated palladium-catalyzed aminocarbonylation reaction that circumvents many of these historical inefficiencies. The core innovation lies in the use of benzisoxazole, which undergoes ring-opening under the catalytic conditions to provide both the nitrogen atom and the carbonyl carbon required for the quinolinone ring formation. This eliminates the need for external carbon monoxide gas, which is notoriously difficult to handle safely on a large scale, and replaces it with solid molybdenum hexacarbonyl as a convenient CO surrogate. The reaction proceeds under relatively mild thermal conditions, typically around 100°C, in the presence of a base like triethylamine and a chiral phosphine ligand such as (S)-BINAP. This setup not only enhances the safety profile of the operation but also improves the atom economy of the transformation. The method demonstrates exceptional versatility, accommodating a wide array of substituents on both the benzisoxazole and the benzyl chloride coupling partners without compromising reaction efficiency. This robustness allows chemists to access complex molecular architectures directly, significantly simplifying the synthetic route and facilitating the rapid production of diverse compound libraries for biological evaluation.
Mechanistic Insights into Palladium-Catalyzed Aminocarbonylation
The success of this synthetic methodology hinges on a carefully orchestrated catalytic cycle involving palladium, molybdenum, and the unique reactivity of the benzisoxazole ring. The reaction initiates with the oxidative addition of the benzyl chloride derivative to the active palladium(0) species generated in situ from palladium acetate and the (S)-BINAP ligand. Simultaneously, molybdenum hexacarbonyl serves as a solid source of carbon monoxide, releasing CO into the reaction medium upon heating, which then coordinates to the palladium center. The benzisoxazole molecule then participates in a nucleophilic attack or insertion sequence, where the N-O bond is cleaved to release the nitrogen atom that eventually becomes part of the lactam ring. This intricate dance of bond breaking and forming is facilitated by the electronic properties of the ligand system, which stabilizes the palladium intermediates and promotes the migratory insertion steps necessary for ring closure. Water is included in the reaction mixture, likely playing a critical role as a proton source to facilitate the final aromatization or hydrolysis steps that yield the stable quinolin-2(1H)one product. Understanding these mechanistic nuances is vital for R&D teams aiming to troubleshoot potential issues during scale-up or when adapting the protocol to novel substrates.
Impurity control is another critical aspect where this mechanism offers distinct advantages over traditional acid-catalyzed cyclizations. Because the reaction relies on a specific transition metal catalytic cycle rather than brute-force thermal activation, side reactions such as polymerization or non-specific decomposition of the starting materials are significantly suppressed. The use of a chiral ligand like (S)-BINAP, although the product itself may not always be chiral at the final stage depending on the substrate, ensures a well-defined coordination sphere around the metal center, promoting the desired pathway over competing off-cycle reactions. The patent data indicates that even with electron-deficient or electron-rich substituents, the reaction maintains high fidelity, producing the target quinolinone with minimal formation of regioisomers or oligomeric byproducts. This high level of selectivity translates directly to simpler downstream processing, as the crude reaction mixtures are cleaner and require less aggressive purification techniques to meet stringent pharmaceutical purity specifications. For quality control laboratories, this means more consistent analytical results and reduced risk of batch rejection due to unknown impurities.
How to Synthesize 3-Arylquinolin-2(1H)-one Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires adherence to specific operational parameters to maximize yield and reproducibility. The process begins with the precise weighing of the catalyst system, typically employing a molar ratio of palladium acetate to ligand of roughly 1:1, alongside 1.5 equivalents of molybdenum hexacarbonyl relative to the limiting reagent. The choice of solvent is also critical, with ethylene glycol dimethyl ether (DME) identified as the optimal medium for solubilizing the reactants while maintaining the stability of the catalytic species. Triethylamine is added in excess, serving as both a base to neutralize the hydrochloric acid byproduct and potentially as a ligand modifier. The reaction is conducted in a sealed vessel to prevent the loss of volatile components and is heated to 100°C for a duration of approximately 26 hours. Detailed standardized synthesis steps see the guide below.
- Combine palladium acetate, (S)-BINAP, molybdenum hexacarbonyl, triethylamine, water, benzisoxazole, and benzyl chloride in DME solvent.
- Heat the reaction mixture to 100°C in a sealed tube and maintain stirring for approximately 26 hours to ensure complete conversion.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target quinolinone derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthetic route presents a compelling value proposition centered on cost efficiency and supply reliability. The primary economic driver is the utilization of benzisoxazole and benzyl chloride derivatives as starting materials, which are commodity chemicals available in bulk quantities from multiple global suppliers. This abundance ensures that the raw material supply chain is robust and less susceptible to the volatility often seen with specialized, custom-synthesized precursors. Furthermore, the elimination of gaseous carbon monoxide removes the need for specialized high-pressure equipment and rigorous safety protocols associated with toxic gas handling, leading to substantial capital expenditure savings and lower operational overheads. The simplified workup procedure, which involves basic filtration and standard chromatography, reduces the consumption of solvents and silica gel compared to multi-step sequences, contributing to a greener and more cost-effective manufacturing process. These factors combine to create a manufacturing profile that is highly attractive for long-term commercial production.
- Cost Reduction in Manufacturing: The economic benefits of this process are derived from the convergence of cheap raw materials and high reaction efficiency. By avoiding expensive reagents and complex protection-deprotection sequences, the overall cost of goods sold (COGS) for the final quinolinone intermediate is significantly lowered. The high yields reported in the patent examples, often exceeding ninety percent for various substrates, mean that less starting material is wasted, directly improving the material balance and reducing the cost per kilogram of the final product. Additionally, the use of a solid CO surrogate like molybdenum hexacarbonyl simplifies logistics and storage compared to managing pressurized gas cylinders, further reducing indirect costs associated with safety compliance and infrastructure maintenance.
- Enhanced Supply Chain Reliability: Supply chain continuity is bolstered by the commercial availability of all key reagents involved in this transformation. Benzisoxazoles and substituted benzyl chlorides are widely produced for various industrial applications, ensuring a steady flow of materials even during market fluctuations. The robustness of the reaction conditions, which tolerate a wide range of functional groups, means that the process is less sensitive to minor variations in raw material quality, reducing the risk of batch failures due to specification drifts. This reliability allows for more accurate production planning and inventory management, enabling manufacturers to meet tight delivery schedules for their clients without the buffer stocks typically required for finicky synthetic processes. Consequently, lead times for high-purity pharmaceutical intermediates can be consistently maintained or even reduced.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, this method aligns well with modern green chemistry principles. The reaction operates at moderate temperatures and does not generate large volumes of corrosive acidic waste, unlike traditional Friedlander or Vilsmeier-Haack protocols. This simplifies wastewater treatment and reduces the environmental footprint of the manufacturing site. The straightforward isolation of the product via filtration and chromatography is easily adaptable to larger scales, where continuous chromatography or crystallization techniques can be implemented to further enhance throughput. The ability to scale this process from gram to kilogram quantities without significant re-optimization makes it an ideal candidate for bridging the gap between early-stage drug discovery and commercial API production, ensuring a smooth transition through the development pipeline.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this palladium-catalyzed synthesis. These insights are derived directly from the experimental data and technical specifications provided in the patent documentation, offering clarity on process capabilities and limitations. Understanding these details helps stakeholders make informed decisions about integrating this technology into their existing manufacturing portfolios. The answers reflect the practical realities of running this chemistry in a production environment, focusing on yield consistency, substrate flexibility, and operational safety.
Q: What is the primary advantage of using benzisoxazole in this synthesis?
A: Benzisoxazole serves a dual function as both the nitrogen source and the formyl (carbonyl) source, eliminating the need for separate carbon monoxide gas or complex formylating agents, thereby simplifying the operational procedure and enhancing safety.
Q: What is the functional group tolerance of this palladium-catalyzed method?
A: The method exhibits excellent tolerance for a wide range of functional groups including halogens (chloro, fluoro), electron-donating groups (methoxy, tert-butyl), and electron-withdrawing groups (cyano, trifluoromethyl), allowing for the synthesis of diverse derivatives without protecting group strategies.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the process utilizes commercially available and inexpensive starting materials, operates under relatively mild thermal conditions (100°C), and employs a straightforward workup procedure involving filtration and chromatography, making it highly amenable to scale-up for industrial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Arylquinolin-2(1H)-one Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient synthetic methodologies like the one described in patent CN113045489B for the future of pharmaceutical manufacturing. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries are successfully translated into viable industrial processes. We are committed to delivering high-purity intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Whether you require custom synthesis of specific quinolinone derivatives or scale-up of existing routes, our facility is designed to handle complex organic transformations with precision and reliability. We understand the critical nature of supply chain security in the pharma industry and strive to be a partner that adds value through technical expertise and operational excellence.
We invite you to engage with our technical procurement team to discuss how this advanced aminocarbonylation technology can benefit your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the potential economic advantages of switching to this novel route for your target molecules. We encourage you to contact us to obtain specific COA data for our catalog compounds or to initiate route feasibility assessments for your proprietary candidates. Let us collaborate to optimize your supply chain and accelerate your drug development timelines with our superior manufacturing capabilities and dedication to quality.