Efficient Palladium-Catalyzed Synthesis of Carbonyl-Bridged Biheterocyclic Compounds for Drug Discovery
Efficient Palladium-Catalyzed Synthesis of Carbonyl-Bridged Biheterocyclic Compounds for Drug Discovery
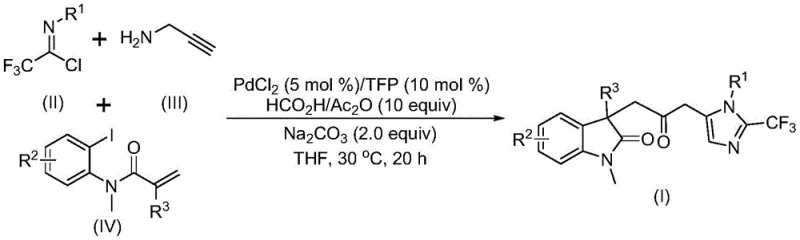
The pharmaceutical industry continuously seeks robust methodologies for constructing complex heterocyclic scaffolds, which serve as the core backbone for numerous bioactive molecules. A significant breakthrough in this domain is detailed in patent CN115353511A, which discloses a novel multi-component method for synthesizing carbonyl-bridged biheterocyclic compounds. This technology represents a paradigm shift in how indolinone and imidazole fused systems are assembled, moving away from hazardous high-pressure carbonylation towards a safer, ambient temperature protocol. By leveraging a palladium-catalyzed cascade reaction, this approach efficiently constructs multiple chemical bonds in a single operational step, addressing critical pain points in process chemistry such as safety, atom economy, and operational simplicity. For R&D teams focused on oncology or CNS drug discovery, where such biheterocyclic motifs are prevalent, this methodology offers a streamlined pathway to access diverse chemical space with high purity and structural integrity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of carbonyl-bridged biheterocyclic frameworks has relied on laborious multi-step sequences or harsh reaction conditions that pose significant challenges for process development. Traditional strategies often involve the direct coupling of two pre-formed heterocyclic substrates, which frequently suffers from low regioselectivity and poor yields due to steric hindrance. Alternatively, oxidative cyclization methods require stoichiometric amounts of expensive oxidants and generate substantial chemical waste, complicating downstream purification and environmental compliance. Furthermore, classical carbonylation reactions typically necessitate the use of toxic carbon monoxide gas under high pressure, requiring specialized autoclave equipment and rigorous safety protocols that increase capital expenditure and operational risk. These limitations collectively result in extended lead times for candidate molecule synthesis and inflated costs, creating bottlenecks in the early stages of drug development pipelines.
The Novel Approach
In stark contrast, the methodology described in CN115353511A introduces a sophisticated yet operationally simple one-pot tandem reaction that elegantly bypasses these traditional hurdles. By utilizing readily available starting materials such as trifluoroethylimidoyl chloride, propargylamine, and acrylamide derivatives, the process achieves the simultaneous formation of C-C, C-N, and C-O bonds through a palladium-catalyzed cascade. Crucially, this innovation replaces dangerous gaseous carbon monoxide with a safe liquid surrogate system comprising formic acid and acetic anhydride, which releases CO in situ under mild conditions. The reaction proceeds efficiently at a温和 temperature of 30°C in common solvents like THF, eliminating the need for energy-intensive heating or high-pressure containment. This not only enhances laboratory safety but also drastically reduces the complexity of the reaction setup, making it highly attractive for both medicinal chemistry campaigns and potential commercial manufacturing.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
Understanding the mechanistic underpinnings of this transformation is vital for R&D directors aiming to optimize reaction parameters or adapt the chemistry to novel substrates. The catalytic cycle is initiated by the oxidative addition of a zero-valent palladium species into the carbon-iodine bond of the acrylamide substrate, generating a reactive organopalladium intermediate. This is followed by an intramolecular Heck-type insertion into the alkene moiety, forming a five-membered palladacycle. Subsequently, the in situ generated carbon monoxide, derived from the decomposition of the formic acid/acetic anhydride mixture, inserts into the palladium-carbon bond to yield an acyl-palladium species. This acyl intermediate is the pivotal junction where the carbonyl bridge is established, setting the stage for the final ring closure. The presence of the trifurylphosphine (TFP) ligand is critical here, as it stabilizes the palladium center and facilitates the migratory insertion steps necessary for high turnover numbers.
Following the carbonylation event, the reaction pathway diverges into a complex intermolecular sequence involving the trifluoroethylimidoyl chloride and propargylamine components. Under the promotion of the base, sodium carbonate, these two reagents undergo an intermolecular carbon-nitrogen bond formation to generate a trifluoroacetamidine intermediate, which subsequently isomerizes. The acyl-palladium species then activates this amidine compound, triggering an intramolecular cyclization that最终 constructs the imidazole ring fused to the indolinone core. This intricate dance of bond formations occurs seamlessly in a single pot, demonstrating remarkable chemoselectivity. The tolerance for diverse functional groups—ranging from electron-withdrawing nitro and trifluoromethyl groups to electron-donating methoxy and alkyl substituents—suggests that the electronic properties of the substrates do not significantly impede the catalytic cycle, ensuring broad applicability across different chemical series.
How to Synthesize Carbonyl-Bridged Biheterocyclic Compounds Efficiently
Implementing this synthesis route requires careful attention to reagent quality and stoichiometry to maximize yield and minimize impurities. The protocol dictates a precise molar ratio of substrates, typically employing an excess of propargylamine and acrylamide relative to the trifluoroethylimidoyl chloride to drive the equilibrium forward. The choice of solvent is also paramount; while aprotic solvents generally support the reaction, tetrahydrofuran (THF) has been identified as the optimal medium for achieving high conversion rates. Post-reaction processing is straightforward, involving simple filtration to remove inorganic salts followed by standard silica gel chromatography, which aligns well with standard operating procedures in most process chemistry laboratories. For detailed procedural specifics regarding reagent addition orders and workup techniques, please refer to the standardized synthesis guide below.
- Prepare the reaction mixture by adding palladium chloride, trifurylphosphine, sodium carbonate, and a mixture of acetic anhydride and formic acid into an organic solvent such as THF.
- Introduce the key substrates: trifluoroethylimidoyl chloride, propargylamine, and the specific acrylamide derivative into the reaction vessel under stirring.
- Maintain the reaction at 30°C for 12 to 20 hours, then proceed with filtration, silica gel mixing, and column chromatography purification to isolate the final product.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this patented technology offers compelling economic and logistical benefits that directly impact the bottom line. The shift from hazardous gas handling to liquid reagents fundamentally alters the risk profile of the manufacturing process, potentially lowering insurance premiums and reducing the need for specialized infrastructure. Moreover, the reliance on commodity chemicals like formic acid, acetic anhydride, and sodium carbonate ensures a stable and cost-effective supply chain, insulating production from the volatility associated with exotic reagents. The high atom economy of the multi-component reaction means less raw material is wasted as byproducts, leading to significant reductions in raw material costs per kilogram of active pharmaceutical ingredient (API) intermediate produced. This efficiency translates directly into improved margins for large-scale manufacturing campaigns.
- Cost Reduction in Manufacturing: The elimination of high-pressure carbon monoxide equipment removes a major capital expenditure barrier, allowing production to occur in standard glass-lined reactors rather than expensive autoclaves. Additionally, the use of inexpensive palladium chloride instead of more exotic ligand-catalyst complexes further drives down catalyst costs. The mild reaction temperature of 30°C significantly reduces energy consumption for heating and cooling compared to traditional reflux conditions, contributing to lower utility costs. By consolidating multiple synthetic steps into a single pot, the process minimizes solvent usage and labor hours associated with intermediate isolations, resulting in substantial overall cost savings for the production of complex biheterocyclic scaffolds.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, including propargylamine and various substituted acrylamides, are widely available from global chemical suppliers, ensuring robust supply continuity. The reaction's tolerance for moisture and air, implied by the use of simple bases and solvents, reduces the stringency of storage and handling requirements, thereby minimizing the risk of batch failures due to environmental exposure. This resilience makes the supply chain more agile and responsive to fluctuating demand from downstream pharmaceutical clients. Furthermore, the ability to synthesize diverse analogs by simply swapping the R-groups on the starting materials allows for rapid response to changing market needs without requalifying entirely new synthetic routes.
- Scalability and Environmental Compliance: The successful demonstration of gram-scale synthesis in the patent data indicates a clear path towards kilogram and ton-scale production without encountering unforeseen exotherms or mixing issues. The avoidance of toxic CO gas simplifies environmental permitting and waste gas treatment processes, aligning with increasingly stringent global environmental regulations. The simplified workup procedure generates less hazardous waste compared to multi-step alternatives, reducing disposal costs and enhancing the sustainability profile of the manufacturing process. This green chemistry advantage is increasingly valued by top-tier pharmaceutical companies seeking to reduce the carbon footprint of their supply chains.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing a reliable basis for decision-making. Understanding these nuances helps stakeholders evaluate the fit of this technology within their existing portfolios and manufacturing capabilities. For further technical clarification or custom feasibility studies, our team is available to provide detailed consultations.
Q: What are the primary advantages of this multi-component synthesis method over traditional coupling reactions?
A: This method utilizes a one-pot tandem cyclization strategy that avoids the need for toxic carbon monoxide gas by using a formic acid/acetic anhydride system. It operates under mild conditions (30°C) and demonstrates excellent functional group tolerance, significantly simplifying the purification process compared to multi-step traditional routes.
Q: Is this synthesis protocol scalable for industrial manufacturing?
A: Yes, the patent explicitly demonstrates the feasibility of scaling the reaction to the gram level with high efficiency. The use of commercially available catalysts like PdCl2 and common solvents like THF supports straightforward scale-up for commercial production of complex pharmaceutical intermediates.
Q: What types of substituents are compatible with this reaction system?
A: The reaction exhibits broad substrate compatibility, tolerating various substituents on the aryl rings including alkyl groups, halogens (chloro, bromo, fluoro), alkoxy groups, trifluoromethyl, and nitro groups. This versatility allows for the rapid generation of diverse compound libraries for SAR studies.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Carbonyl-Bridged Biheterocyclic Compounds Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this palladium-catalyzed multi-component synthesis in accelerating drug discovery programs. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop to plant floor is seamless and efficient. Our state-of-the-art facilities are equipped to handle sensitive palladium chemistry with stringent purity specifications, supported by rigorous QC labs that guarantee every batch meets the highest international standards. We understand that consistency and quality are non-negotiable in the pharmaceutical supply chain, and our dedicated process chemistry team is committed to optimizing this route for maximum yield and minimal impurity profiles.
We invite you to collaborate with us to leverage this advanced technology for your next project. By partnering with NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and target timelines. Our technical procurement team is ready to discuss your needs in detail and can provide specific COA data and route feasibility assessments upon request. Let us help you navigate the complexities of modern chemical synthesis, delivering high-quality intermediates that empower your innovation and drive your business forward.