Advanced Pd-Catalyzed Synthesis of Carbonyl-Bridged Biheterocyclic Compounds for Commercial Pharmaceutical Applications
Advanced Pd-Catalyzed Synthesis of Carbonyl-Bridged Biheterocyclic Compounds for Commercial Pharmaceutical Applications
The pharmaceutical industry continuously seeks efficient pathways to construct complex heterocyclic scaffolds, which serve as the core backbone for numerous bioactive molecules. Patent CN115353511A introduces a groundbreaking multi-component methodology for synthesizing carbonyl-bridged biheterocyclic compounds, specifically targeting the fusion of indolinone and imidazole motifs. This innovation addresses critical challenges in modern organic synthesis by eliminating the need for hazardous carbon monoxide gas while maintaining high reaction efficiency and substrate versatility. For R&D directors and process chemists, this represents a significant leap forward in accessing diversified chemical space for drug discovery programs. The technology leverages a sophisticated palladium-catalyzed cascade reaction that operates under remarkably mild conditions, typically around 30°C, ensuring the integrity of sensitive functional groups often present in advanced intermediates.
From a supply chain perspective, the reliance on readily available starting materials such as trifluoroethylimidoyl chloride, propargylamine, and acrylamide derivatives simplifies procurement logistics. The ability to generate these high-value structures in a single pot reduces the number of isolation steps, thereby minimizing waste generation and processing time. This aligns perfectly with the industry's shift towards greener chemistry and sustainable manufacturing practices. Furthermore, the patent highlights the successful expansion of this protocol to gram-scale reactions, providing strong evidence for its potential in commercial scale-up of complex pharmaceutical intermediates. As a reliable pharmaceutical intermediate supplier, understanding such proprietary technologies allows us to offer clients superior route feasibility assessments and cost-effective production strategies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of biheterocyclic systems has relied on three primary strategies, each fraught with significant limitations that hinder industrial application. The first approach involves the direct coupling of two pre-formed heterocyclic substrates, which often suffers from low atom economy and requires expensive, pre-functionalized building blocks. The second strategy utilizes oxidative cyclization of substrates containing dual nucleophiles, a process that frequently demands harsh oxidizing agents and elevated temperatures, leading to poor functional group tolerance and safety concerns. The third conventional method employs transition metal-catalyzed tandem cyclizations, which, while efficient, traditionally struggle when applied to carbonyl-bridged systems due to the difficulty of introducing the carbonyl moiety safely and efficiently.
Specifically, traditional carbonylation reactions often necessitate the use of high-pressure carbon monoxide gas, posing severe safety risks and requiring specialized, costly equipment that many contract manufacturing organizations lack. Additionally, these older methods frequently exhibit narrow substrate scopes, failing to accommodate diverse substituents like halogens or electron-withdrawing groups without significant yield erosion. The multi-step nature of many legacy syntheses also results in accumulated impurities, complicating downstream purification and increasing the overall cost of goods. These factors collectively create bottlenecks in the development of new drug candidates, necessitating a more robust and flexible synthetic solution.
The Novel Approach
The methodology disclosed in CN115353511A revolutionizes this landscape by employing a palladium-catalyzed multi-component reaction that seamlessly integrates carbonylation and cyclization in a single operation. By utilizing a mixture of formic acid and acetic anhydride as a safe and effective carbon monoxide surrogate, the process completely bypasses the need for toxic CO gas cylinders and high-pressure reactors. This not only enhances laboratory safety but also drastically simplifies the engineering requirements for scaling the reaction to pilot and commercial plants. The reaction proceeds smoothly at a mild temperature of 30°C, preserving the stability of thermally labile groups and enabling the synthesis of a wide array of substituted derivatives.
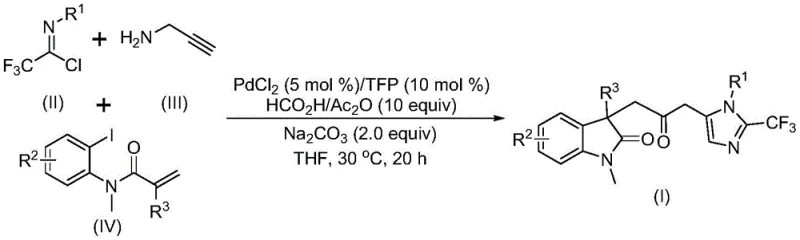
This novel approach demonstrates exceptional substrate compatibility, successfully accommodating various substituents on the aromatic rings, including methyl, methoxy, chloro, bromo, trifluoromethyl, and nitro groups. The one-pot nature of the reaction means that three distinct chemical bonds are formed simultaneously, maximizing atom economy and reducing solvent consumption. For procurement managers, this translates to cost reduction in API manufacturing by lowering raw material costs and minimizing waste disposal fees. The high efficiency and yield reported in the patent examples suggest that this route is not merely a laboratory curiosity but a viable candidate for industrial adoption, offering a streamlined path to valuable indolinone-imidazole hybrids.
Mechanistic Insights into Palladium-Catalyzed Carbonylation Cascade
The mechanistic pathway of this transformation is a testament to the elegance of modern organometallic catalysis, involving a intricate sequence of oxidative addition, insertion, and cyclization events. The cycle likely initiates with the oxidative addition of a zero-valent palladium species into the carbon-iodine bond of the acrylamide substrate, generating a reactive aryl-palladium intermediate. This is followed by an intramolecular Heck-type reaction, where the palladium center inserts into the alkene moiety, forming a divalent alkyl-palladium species that sets the stage for ring closure. Subsequently, the carbon monoxide generated in situ from the formic acid/acetic anhydride mixture coordinates to the palladium center, facilitating a carbonylation step that yields a crucial acyl-palladium intermediate.
Parallel to the palladium cycle, the base-promoted reaction between trifluoroethylimidoyl chloride and propargylamine generates a trifluoroacetamidine intermediate, which undergoes isomerization to become the active nucleophile. The acyl-palladium species then activates this amidine compound, triggering an intramolecular cyclization that constructs the imidazole ring while simultaneously releasing the palladium catalyst to re-enter the cycle. This dual-activation mechanism ensures high regioselectivity and minimizes side reactions, resulting in the clean formation of the carbonyl-bridged biheterocyclic core. Understanding these mechanistic nuances is vital for R&D teams aiming to optimize reaction parameters or adapt the chemistry to novel substrates.
Impurity control is inherently built into this mechanism due to the high specificity of the palladium catalyst and the mild reaction conditions. The use of tri-2-furylphosphine (TFP) as a ligand stabilizes the palladium center, preventing the formation of palladium black and ensuring consistent catalytic activity throughout the 12 to 20-hour reaction window. Furthermore, the choice of sodium carbonate as a base provides sufficient alkalinity to promote the amidine formation without causing hydrolysis of the sensitive imidoyl chloride or ester functionalities. This careful balance of reagents results in a crude reaction profile that is amenable to standard purification techniques, such as silica gel column chromatography, yielding products with high purity suitable for biological testing.
How to Synthesize Carbonyl-Bridged Biheterocyclic Compounds Efficiently
The practical execution of this synthesis is designed for simplicity and reproducibility, making it accessible for both academic research and industrial process development. The protocol involves charging a reaction vessel with the palladium catalyst, ligand, base, and the CO source mixture in an aprotic solvent like tetrahydrofuran (THF). To this mixture, the three key substrates are added in specific molar ratios, typically with a slight excess of propargylamine and acrylamide to drive the reaction to completion. The reaction is then stirred at 30°C for a period ranging from 12 to 20 hours, after which standard workup procedures involving filtration and chromatographic purification afford the target biheterocyclic compounds.
- Combine palladium chloride catalyst, TFP ligand, sodium carbonate base, and the formic acid/acetic anhydride CO source in an organic solvent like THF.
- Add the three key substrates: trifluoroethylimidoyl chloride, propargylamine, and the acrylamide derivative to the reaction mixture under stirring.
- Maintain the reaction at 30°C for 12 to 20 hours, followed by filtration, silica gel treatment, and column chromatography purification to isolate the final product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented technology offers tangible benefits that extend beyond mere chemical novelty. The primary advantage lies in the substantial cost savings achieved through the use of commodity chemicals as starting materials. Trifluoroethylimidoyl chloride, propargylamine, and acrylamide derivatives are commercially available in bulk quantities, reducing the risk of supply chain disruptions associated with exotic or custom-synthesized reagents. Moreover, the elimination of high-pressure carbon monoxide equipment removes a significant capital expenditure barrier, allowing for faster deployment of production lines and reduced overhead costs.
- Cost Reduction in Manufacturing: The one-pot nature of this multi-component reaction significantly reduces the number of unit operations required compared to stepwise synthesis. By combining bond-forming events, manufacturers can save on solvent usage, energy consumption for heating and cooling, and labor hours associated with intermediate isolations. The mild reaction temperature of 30°C further contributes to energy efficiency, as it does not require intensive heating or cryogenic cooling systems. These cumulative efficiencies lead to a lower cost of goods sold (COGS), enhancing the competitiveness of the final pharmaceutical product in the global market.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route against varying substrate electronics ensures a stable supply of diverse intermediates. Since the method tolerates a wide range of functional groups, including halogens and nitro groups, it allows for the late-stage diversification of molecules without needing to redesign the entire synthetic pathway. This flexibility is crucial for responding to changing market demands or regulatory requirements during drug development. Additionally, the use of standard solvents like THF and common bases like sodium carbonate ensures that raw materials can be sourced from multiple vendors, mitigating the risk of single-source dependency.
- Scalability and Environmental Compliance: The patent data indicates successful gram-scale synthesis, suggesting a clear path to kilogram and ton-scale production. The absence of toxic gases and the use of relatively benign reagents simplify waste management and environmental compliance, aligning with increasingly stringent global regulations on chemical manufacturing. The simplified post-treatment process, involving basic filtration and chromatography, reduces the generation of hazardous waste streams. This environmental friendliness not only lowers disposal costs but also enhances the corporate sustainability profile of the manufacturing entity, a key metric for modern pharmaceutical partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing a clear picture of what partners can expect when adopting this methodology. Understanding these details helps in making informed decisions about process integration and resource allocation.
Q: What are the primary advantages of this Pd-catalyzed method over traditional biheterocycle synthesis?
A: Unlike conventional methods that often require harsh conditions or toxic carbon monoxide gas, this patented process utilizes a safe formic acid/acetic anhydride system as a CO surrogate at mild temperatures (30°C), significantly improving operational safety and functional group tolerance.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the patent explicitly demonstrates the method's scalability to gram-level reactions with high efficiency. The use of commercially available starting materials and standard purification techniques like column chromatography supports its feasibility for commercial scale-up of complex pharmaceutical intermediates.
Q: What is the substrate scope regarding the R groups in the final biheterocyclic structure?
A: The method exhibits excellent substrate compatibility, tolerating various substituents such as alkyl, alkoxy, halogens (Cl, Br, F), trifluoromethyl, and nitro groups on the aromatic rings, allowing for the diverse design of drug-like molecules.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Carbonyl-Bridged Biheterocyclic Compounds Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the Pd-catalyzed multi-component synthesis described in CN115353511A for the development of next-generation therapeutics. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from laboratory bench to manufacturing plant is seamless and efficient. We are committed to delivering high-purity intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify every batch.
We invite you to collaborate with us to leverage this advanced chemistry for your drug discovery pipelines. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific project needs. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can accelerate your timeline and optimize your budget. Let us be your partner in turning complex chemical challenges into commercial successes.